
Is Finding Out Your Baby’s Genetics A New Responsibility of Parenting?
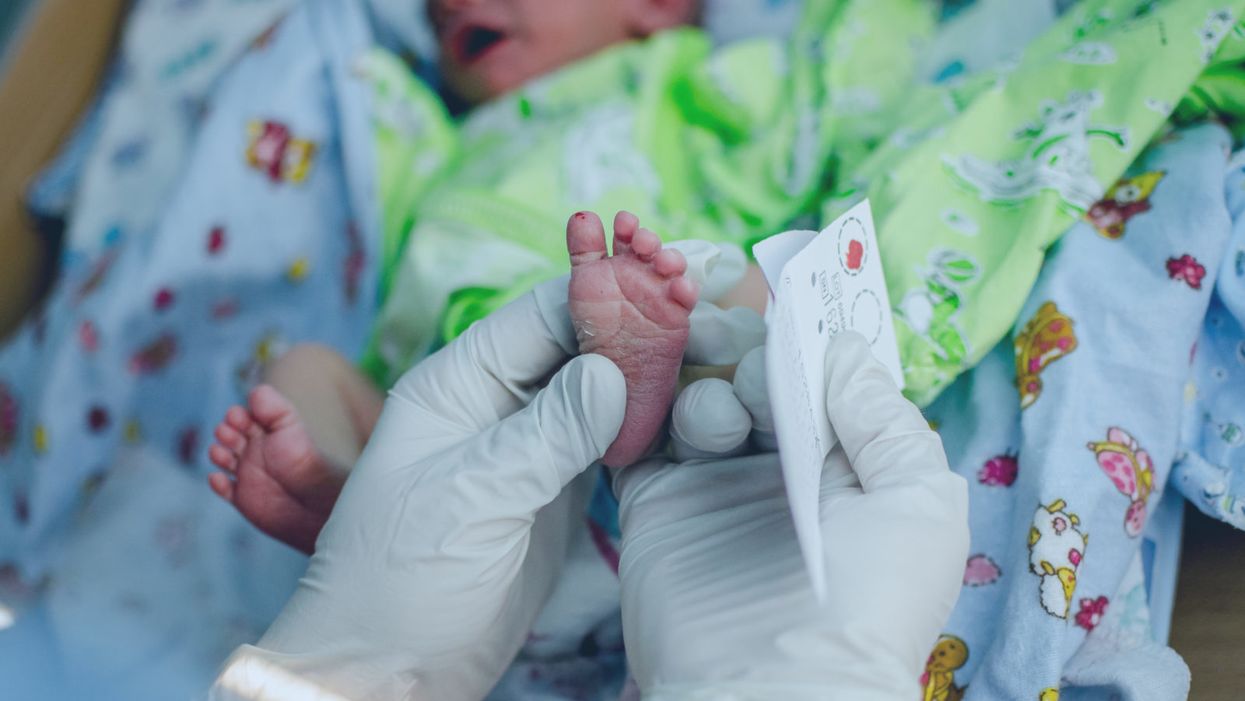
A doctor pricks the heel of a newborn for a blood test.
Hours after a baby is born, its heel is pricked with a lancet. Drops of the infant's blood are collected on a porous card, which is then mailed to a state laboratory. The dried blood spots are screened for around thirty conditions, including phenylketonuria (PKU), the metabolic disorder that kick-started this kind of newborn screening over 60 years ago. In the U.S., parents are not asked for permission to screen their child. Newborn screening programs are public health programs, and the assumption is that no good parent would refuse a screening test that could identify a serious yet treatable condition in their baby.
Learning as much as you can about your child's health might seem like a natural obligation of parenting. But it's an assumption that I think needs to be much more closely examined.
Today, with the introduction of genome sequencing into clinical medicine, some are asking whether newborn screening goes far enough. As the cost of sequencing falls, should parents take a more expansive look at their children's health, learning not just whether they have a rare but treatable childhood condition, but also whether they are at risk for untreatable conditions or for diseases that, if they occur at all, will strike only in adulthood? Should genome sequencing be a part of every newborn's care?
It's an idea that appeals to Anne Wojcicki, the founder and CEO of the direct-to-consumer genetic testing company 23andMe, who in a 2016 interview with The Guardian newspaper predicted that having newborns tested would soon be considered standard practice—"as critical as testing your cholesterol"—and a new responsibility of parenting. Wojcicki isn't the only one excited to see everyone's genes examined at birth. Francis Collins, director of the National Institutes of Health and perhaps the most prominent advocate of genomics in the United States, has written that he is "almost certain … that whole-genome sequencing will become part of new-born screening in the next few years." Whether that would happen through state-mandated screening programs, or as part of routine pediatric care—or perhaps as a direct-to-consumer service that parents purchase at birth or receive as a baby-shower gift—is not clear.
Learning as much as you can about your child's health might seem like a natural obligation of parenting. But it's an assumption that I think needs to be much more closely examined, both because the results that genome sequencing can return are more complex and more uncertain than one might expect, and because parents are not actually responsible for their child's lifelong health and well-being.
What is a parent supposed to do about such a risk except worry?
Existing newborn screening tests look for the presence of rare conditions that, if identified early in life, before the child shows any symptoms, can be effectively treated. Sequencing could identify many of these same kinds of conditions (and it might be a good tool if it could be targeted to those conditions alone), but it would also identify gene variants that confer an increased risk rather than a certainty of disease. Occasionally that increased risk will be significant. About 12 percent of women in the general population will develop breast cancer during their lives, while those who have a harmful BRCA1 or BRCA2 gene variant have around a 70 percent chance of developing the disease. But for many—perhaps most—conditions, the increased risk associated with a particular gene variant will be very small. Researchers have identified over 600 genes that appear to be associated with schizophrenia, for example, but any one of those confers only a tiny increase in risk for the disorder. What is a parent supposed to do about such a risk except worry?
Sequencing results are uncertain in other important ways as well. While we now have the ability to map the genome—to create a read-out of the pairs of genetic letters that make up a person's DNA—we are still learning what most of it means for a person's health and well-being. Researchers even have a name for gene variants they think might be associated with a disease or disorder, but for which they don't have enough evidence to be sure. They are called "variants of unknown (or uncertain) significance (VUS), and they pop up in most people's sequencing results. In cancer genetics, where much research has been done, about 1 in 5 gene variants are reclassified over time. Most are downgraded, which means that a good number of VUS are eventually designated benign.
While one parent might reasonably decide to learn about their child's risk for a condition about which nothing can be done medically, a different, yet still thoroughly reasonable, parent might prefer to remain ignorant so that they can enjoy the time before their child is afflicted.
Then there's the puzzle of what to do about results that show increased risk or even certainty for a condition that we have no idea how to prevent. Some genomics advocates argue that even if a result is not "medically actionable," it might have "personal utility" because it allows parents to plan for their child's future needs, to enroll them in research, or to connect with other families whose children carry the same genetic marker.
Finding a certain gene variant in one child might inform parents' decisions about whether to have another—and if they do, about whether to use reproductive technologies or prenatal testing to select against that variant in a future child. I have no doubt that for some parents these personal utility arguments are persuasive, but notice how far we've now strayed from the serious yet treatable conditions that motivated governments to set up newborn screening programs, and to mandate such testing for all.
Which brings me to the other problem with the call for sequencing newborn babies: the idea that even if it's not what the law requires, it's what good parents should do. That idea is very compelling when we're talking about sequencing results that show a serious threat to the child's health, especially when interventions are available to prevent or treat that condition. But as I have shown, many sequencing results are not of this type.
While one parent might reasonably decide to learn about their child's risk for a condition about which nothing can be done medically, a different, yet still thoroughly reasonable, parent might prefer to remain ignorant so that they can enjoy the time before their child is afflicted. This parent might decide that the worry—and the hypervigilence it could inspire in them—is not in their child's best interest, or indeed in their own. This parent might also think that it should be up to the child, when he or she is older, to decide whether to learn about his or her risk for adult-onset conditions, especially given that many adults at high familial risk for conditions like Alzheimer's or Huntington's disease choose never to be tested. This parent will value the child's future autonomy and right not to know more than they value the chance to prepare for a health risk that won't strike the child until 40 or 50 years in the future.
Parents are not obligated to learn about their children's risk for a condition that cannot be prevented, has a small risk of occurring, or that would appear only in adulthood.
Contemporary understandings of parenting are famously demanding. We are asked to do everything within our power to advance our children's health and well-being—to act always in our children's best interests. Against that backdrop, the need to sequence every newborn baby's genome might seem obvious. But we should be skeptical. Many sequencing results are complex and uncertain. Parents are not obligated to learn about their children's risk for a condition that cannot be prevented, has a small risk of occurring, or that would appear only in adulthood. To suggest otherwise is to stretch parental responsibilities beyond the realm of childhood and beyond factors that parents can control.
Podcast: Trusting Science with Dr. Sudip Parikh, CEO of AAAS

The "Making Sense of Science" podcast features interviews with leading experts about health innovations and the big ethical and social questions they raise. The podcast is hosted by Matt Fuchs, editor of the award-winning science outlet Leaps.org.
As Pew research showed last month, many Americans have less confidence in science these days - our collective trust has declined to levels below when the pandemic began. But leaders like Dr. Sudip Parikh are taking important steps to more fully engage people in scientific progress, including breakthroughs that could benefit health and prevent disease. In January 2020, Sudip became the 19th Chief Executive Officer of the American Association for the Advancement of Science (AAAS), an international nonprofit that seeks to advance science, engineering and innovation throughout the world, with 120,000 members in 91 countries. He is the executive publisher of Science, one of the top academic journals in the world, and the Science family of journals.
Listen to the episode
Listen on Apple | Listen on Spotify | Listen on Stitcher | Listen on Amazon | Listen on Google
In this episode, Sudip and I talk about:
- Reasons to be excited about health innovations that could come to fruition in the next several years.
- Sudip's thoughts about areas of health innovation where we should be especially cautious.
- Strategies for scientists and journalists to instill greater trust in science.
- How to tap into and nurture kids' passion for STEM subjects.
- The best roles for experts to play in society and the challenges they face.
And we pack several other fascinating topics into our 35 minutes. Here are links to check out and learn more about Sudip Parikh and AAAS:
- Sudip Parikh's official bio - https://www.aaas.org/person/sudip-parikh
- Sudip Parikh, Why We Must Rebuild Trust in Science, Trend Magazine, Feb. 9, 2021 - https://www.pewtrusts.org/en/trend/archive/winter-...
- Follow Sudip on Twitter - https://twitter.com/sudipsparikh
- AAAS website - https://www.aaas.org/
- AAAS podcast - https://www.science.org/podcasts
- The latest issue of Science - https://www.science.org/
- Science Journals homepage - https://www.science.org/journals
- AAAS Mentor Resources - https://www.aaas.org/stemmentoring
- AAAS Science Journalism Awards - https://sjawards.aaas.org/enter
- Pew Research Center Report, Americans' Trust in Scientists, Other Groups Declines, Feb. 15, 2022 https://www.pewresearch.org/science/2022/02/15/ame...

An implant, combined with the glasses and tiny video camera modeled in this photo, could improve the eyesight of millions of people with degenerative eye diseases in the coming years.
For millions of people with macular degeneration, treatment options are slim. The disease causes loss of central vision, which allows us to see straight ahead, and is highly dependent on age, with people over 75 at approximately 30% risk of developing the disorder. The BrightFocus Foundation estimates 11 million people in the U.S. currently have one of three forms of the disease.
Recently, ophthalmologists including Daniel Palanker at Stanford University published research showing advances in the PRIMA retinal implant, which could help people with advanced, age-related macular degeneration regain some of their sight. In a feasibility study, five patients had a pixelated chip implanted behind the retina, and three were able to see using their remaining peripheral vision and—thanks to the implant—their partially restored central vision at the same time.
Should people with macular degeneration be excited about these results?
“Every week, if not every day, patients come to me with this question because it's devastating when they lose their central vision,” says retinal surgeon Lynn Huang. About 40% of her patients have macular degeneration. Huang tells them that these implants, along with new medications and stem cell therapies, could be useful in the coming years.
“The goal here is to replace the missing photoreceptors with photovoltaic pixels, basically like little solar panels,” Palanker says.
That implant, a pixelated chip, works together with a tiny video camera on a specially designed pair of eyeglasses, which can be adjusted for each patient’s prescription. The video camera relays processed images to the chip, which electrically stimulates inner retinal neurons. These neurons, in turn, relay information to the brain’s visual cortex through the optic nerve. The chip restores patients’ central sight, but not completely. The artificial vision is basically monochromatic (whitish-yellowish) and fairly blurry; patients were still legally blind even after the implant, except when using a zoom function on the camera, but those with proper chip placement could make out large letters.
“The goal here is to replace the missing photoreceptors with photovoltaic pixels, basically like little solar panels,” Palanker says. These pixels, located on the implanted chip, convert light into pulsed electrical currents that stimulate retinal neurons. In time, Palanker hopes to improve the chips, resulting in bigger boosts to visual acuity.
The pixelated chips are surgically implanted during a process Palanker admits is still “a surgical learning curve.” In the study, three chips were implanted correctly, one was placed incorrectly, and another patient’s chip moved after the procedure; he did not follow post-surgical recommendations. One patient passed away during the study for unrelated reasons.
University of Maryland retinal specialist Kenneth Taubenslag, who was not involved in the study, said that subretinal surgeries have become less common in recent years, but expects implants to spur improvements in these techniques. “I think as people get more experience, [they’ll] probably get more reliable placement of the implant,” he said, pointing out that even the patient with the misplaced chip was able to gain some light perception, if not the same visual acuity as other patients.
Retinal implants have come under scrutiny lately. IEEE Spectrum reported that Second Sight, manufacturer of the Argus II implant used for people with retinitis pigmentosa, a genetic disease that causes vision loss, would no longer support the product. After selling hundreds of the implants at $150,000 apiece, company leaders announced they’d “decided to pursue an orderly wind down” of Second Sight in March 2020 in the wake of financial issues. Last month, the company announced a merger, shifting its focus to a new retinal implant, raising questions for patients who have Argus II implants.
Retinal surgeon Eugene de Juan of the University of California, San Francisco, was involved with early studies of the Argus implants, though his participation ended over a decade ago, before the device was marketed by Second Sight. He says he would consider recommending future implants to patients with macular degeneration, given the promise of the technology and the lack of other alternatives.
“I tell my patients that this is an area of active research and development, and it's getting better and better, so let's not give up hope,” de Juan says. He believes cautious optimism for Palanker’s implant is appropriate: “It's not the first, it's not the only, but it's a good approach with a good team.”