
Is Finding Out Your Baby’s Genetics A New Responsibility of Parenting?
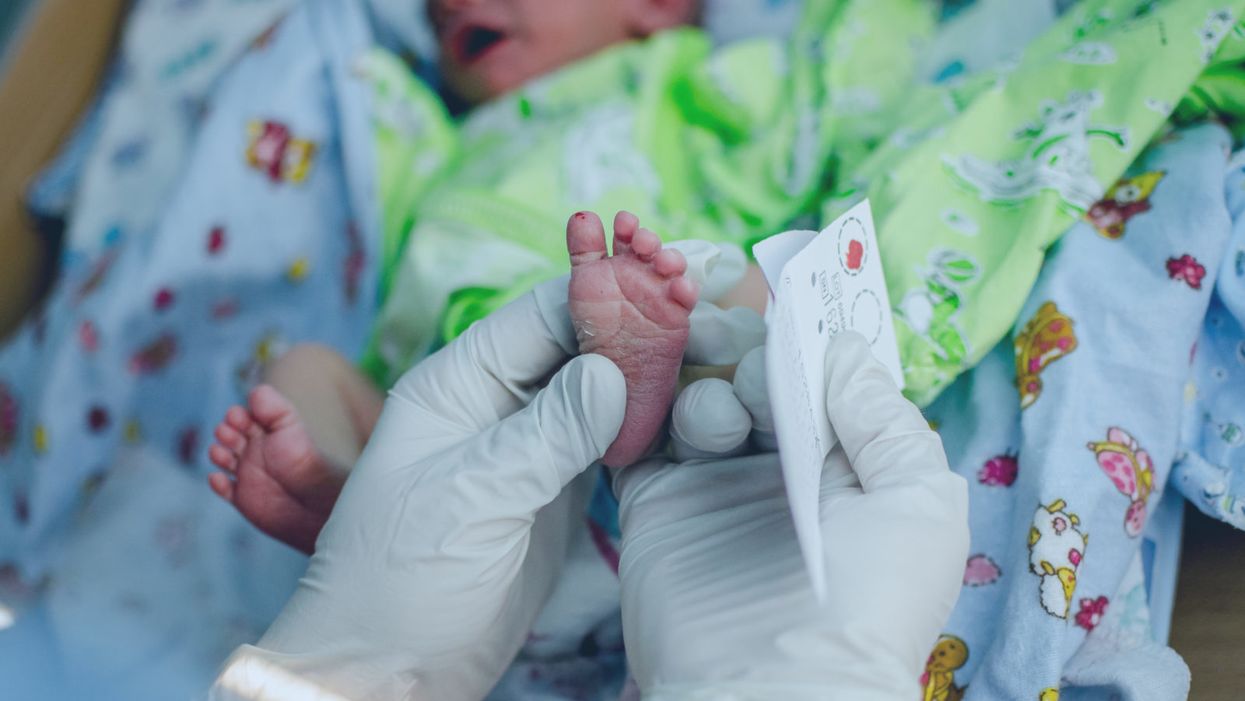
A doctor pricks the heel of a newborn for a blood test.
Hours after a baby is born, its heel is pricked with a lancet. Drops of the infant's blood are collected on a porous card, which is then mailed to a state laboratory. The dried blood spots are screened for around thirty conditions, including phenylketonuria (PKU), the metabolic disorder that kick-started this kind of newborn screening over 60 years ago. In the U.S., parents are not asked for permission to screen their child. Newborn screening programs are public health programs, and the assumption is that no good parent would refuse a screening test that could identify a serious yet treatable condition in their baby.
Learning as much as you can about your child's health might seem like a natural obligation of parenting. But it's an assumption that I think needs to be much more closely examined.
Today, with the introduction of genome sequencing into clinical medicine, some are asking whether newborn screening goes far enough. As the cost of sequencing falls, should parents take a more expansive look at their children's health, learning not just whether they have a rare but treatable childhood condition, but also whether they are at risk for untreatable conditions or for diseases that, if they occur at all, will strike only in adulthood? Should genome sequencing be a part of every newborn's care?
It's an idea that appeals to Anne Wojcicki, the founder and CEO of the direct-to-consumer genetic testing company 23andMe, who in a 2016 interview with The Guardian newspaper predicted that having newborns tested would soon be considered standard practice—"as critical as testing your cholesterol"—and a new responsibility of parenting. Wojcicki isn't the only one excited to see everyone's genes examined at birth. Francis Collins, director of the National Institutes of Health and perhaps the most prominent advocate of genomics in the United States, has written that he is "almost certain … that whole-genome sequencing will become part of new-born screening in the next few years." Whether that would happen through state-mandated screening programs, or as part of routine pediatric care—or perhaps as a direct-to-consumer service that parents purchase at birth or receive as a baby-shower gift—is not clear.
Learning as much as you can about your child's health might seem like a natural obligation of parenting. But it's an assumption that I think needs to be much more closely examined, both because the results that genome sequencing can return are more complex and more uncertain than one might expect, and because parents are not actually responsible for their child's lifelong health and well-being.
What is a parent supposed to do about such a risk except worry?
Existing newborn screening tests look for the presence of rare conditions that, if identified early in life, before the child shows any symptoms, can be effectively treated. Sequencing could identify many of these same kinds of conditions (and it might be a good tool if it could be targeted to those conditions alone), but it would also identify gene variants that confer an increased risk rather than a certainty of disease. Occasionally that increased risk will be significant. About 12 percent of women in the general population will develop breast cancer during their lives, while those who have a harmful BRCA1 or BRCA2 gene variant have around a 70 percent chance of developing the disease. But for many—perhaps most—conditions, the increased risk associated with a particular gene variant will be very small. Researchers have identified over 600 genes that appear to be associated with schizophrenia, for example, but any one of those confers only a tiny increase in risk for the disorder. What is a parent supposed to do about such a risk except worry?
Sequencing results are uncertain in other important ways as well. While we now have the ability to map the genome—to create a read-out of the pairs of genetic letters that make up a person's DNA—we are still learning what most of it means for a person's health and well-being. Researchers even have a name for gene variants they think might be associated with a disease or disorder, but for which they don't have enough evidence to be sure. They are called "variants of unknown (or uncertain) significance (VUS), and they pop up in most people's sequencing results. In cancer genetics, where much research has been done, about 1 in 5 gene variants are reclassified over time. Most are downgraded, which means that a good number of VUS are eventually designated benign.
While one parent might reasonably decide to learn about their child's risk for a condition about which nothing can be done medically, a different, yet still thoroughly reasonable, parent might prefer to remain ignorant so that they can enjoy the time before their child is afflicted.
Then there's the puzzle of what to do about results that show increased risk or even certainty for a condition that we have no idea how to prevent. Some genomics advocates argue that even if a result is not "medically actionable," it might have "personal utility" because it allows parents to plan for their child's future needs, to enroll them in research, or to connect with other families whose children carry the same genetic marker.
Finding a certain gene variant in one child might inform parents' decisions about whether to have another—and if they do, about whether to use reproductive technologies or prenatal testing to select against that variant in a future child. I have no doubt that for some parents these personal utility arguments are persuasive, but notice how far we've now strayed from the serious yet treatable conditions that motivated governments to set up newborn screening programs, and to mandate such testing for all.
Which brings me to the other problem with the call for sequencing newborn babies: the idea that even if it's not what the law requires, it's what good parents should do. That idea is very compelling when we're talking about sequencing results that show a serious threat to the child's health, especially when interventions are available to prevent or treat that condition. But as I have shown, many sequencing results are not of this type.
While one parent might reasonably decide to learn about their child's risk for a condition about which nothing can be done medically, a different, yet still thoroughly reasonable, parent might prefer to remain ignorant so that they can enjoy the time before their child is afflicted. This parent might decide that the worry—and the hypervigilence it could inspire in them—is not in their child's best interest, or indeed in their own. This parent might also think that it should be up to the child, when he or she is older, to decide whether to learn about his or her risk for adult-onset conditions, especially given that many adults at high familial risk for conditions like Alzheimer's or Huntington's disease choose never to be tested. This parent will value the child's future autonomy and right not to know more than they value the chance to prepare for a health risk that won't strike the child until 40 or 50 years in the future.
Parents are not obligated to learn about their children's risk for a condition that cannot be prevented, has a small risk of occurring, or that would appear only in adulthood.
Contemporary understandings of parenting are famously demanding. We are asked to do everything within our power to advance our children's health and well-being—to act always in our children's best interests. Against that backdrop, the need to sequence every newborn baby's genome might seem obvious. But we should be skeptical. Many sequencing results are complex and uncertain. Parents are not obligated to learn about their children's risk for a condition that cannot be prevented, has a small risk of occurring, or that would appear only in adulthood. To suggest otherwise is to stretch parental responsibilities beyond the realm of childhood and beyond factors that parents can control.
Patients voice hope and relief as FDA gives third-ever drug approval for ALS

On Sept. 29, the FDA approved Relyvrio, a new drug for ALS, even though a study of 137 ALS patients did not result in “substantial evidence” that Relyvrio was effective.
At age 52, Glen Rouse suffered from arm weakness and a lot of muscle twitches. “I first thought something was wrong when I could not throw a 50-pound bag of dog food over the tailgate of my truck—something I use to do effortlessly,” said the 54-year-old resident of Anderson, California, about three hours north of San Francisco.
In August, Rouse retired as a forester for a private timber company, a job he had held for 31 years. The impetus: amyotrophic lateral sclerosis, or ALS, a progressive neuromuscular disease that is commonly known as Lou Gehrig’s disease, named after the New York Yankees’ first baseman who succumbed to it less than a month shy of his 38th birthday in 1941. ALS eventually robs an individual of the ability to talk, walk, chew, swallow and breathe.
Rouse is now dependent on ventilation through a nasal mask and uses a powerchair to get around. “I can no longer walk or use my arms very well,” he said. “I can still move my wrists and fingers. I can also transfer from my chair to the toilet if I have two of my friends help me.”
It’s “shocking” that modern medicine has very little to offer to people with this devastating condition, Rouse said. But there is hope on the horizon. Yesterday, the U.S. Food and Drug Administration approved Relyvrio, a drug made up of two parts, sodium phenylbutyrate and taurursodiol, to treat patients with ALS.
“This approval provides another important treatment option for ALS, a life-threatening disease that currently has no cure,” said Billy Dunn, director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, in a statement. “The FDA remains committed to facilitating the development of additional ALS treatments.”
Until this point, the FDA had approved only two other medications—Riluzole (rilutek) in 1995 and Radicava (edaravone) in 2017—to extend life in patients with ALS, which typically kills within two to five years after diagnosis. That’s why earlier this week, Rouse was optimistic about the FDA’s likely approval of a controversial new drug for ALS.
When Relyvrio is taken in addition to Riluzole, it appears to slow functional decline by an additional 25 percent and extend life by another 6 to 10 months, said Richard Bedlak, director of the Duke ALS Clinic. “It is not a cure, but it is definitely a step forward.”
“The whole ALS community is extremely excited about it,” he said the day before Relyvrio’s expected approval. “We are very hopeful. We’re on pins and needles.”
A study of 137 ALS patients did not result in “substantial evidence” that Relyvrio was effective, the agency’s Peripheral and Central Nervous System Drugs Advisory Committee concluded in March. However, after some persuasion from FDA officials, patients and their families, the committee met again and decided to recommend approving the drug.
In January 2019, following an ALS diagnosis at age 58 in October the previous year, Jeff Sarnacki, of Chester, Maryland, was accepted into a trial for Relyvrio. “Because of the trial, we did experience hope and a greater sense of help than had we not had that opportunity,” said Juliet Taylor, his wife and caregiver. They both believed the drug “worked for him in giving him more time.”
In June 2019, Sarnacki chose an open-label extension, offered to patients by drug researchers after a study ends, and took the active drug until he died peacefully at home under hospice care in May 2020, five days after his 60th birthday. A retired agent with the federal Bureau of Alcohol, Tobacco, Firearms and Explosives who later worked as a security consultant, Sarnacki lived about 19 months after diagnosis, which is shorter than the typical prognosis.
His symptoms began with leg cramps in fall 2017 and foot drop in early 2018. A feeding tube was placed in 2019, as it became necessary early in his illness, Taylor said. He also took Radicava and Riluzole, the two previously approved drugs, for his ALS. “We were both incredulous that, so many years after Lou Gehrig’s own diagnosis, there were so few treatments available,” she said.
The dearth of successful treatments for ALS is “certainly not for lack of trying,” said Karen Raley Steffens, a registered nurse and ALS support services coordinator at the Les Turner ALS Foundation in Skokie, Ill. “There are thousands of researchers and scientists all over the world working tirelessly to try to develop treatments for ALS.”
Unfortunately, she added, research takes time and exorbitant amounts of funding, while bureaucratic challenges persist. The rare disease also manifests and progresses in many different ways, so many treatments are needed.
As of 2017, the Centers for Disease Control and Prevention estimated that more than 31,000 people in the U.S. live with ALS, and an average of 5,000 people are newly diagnosed every year. It is slightly more common in men than women. Most people are diagnosed between the ages of 55 and 75.
Most cases of ALS are sporadic, meaning that doctors don’t know the cause. There is about a one-year interval between symptom onset and an ALS diagnosis for most patients, so many motor neurons are lost by the time individuals can enroll in a clinical trial, said Richard Bedlack, professor of neurology and director of the Duke ALS Clinic in Durham, North Carolina.
Bedlack found the new drug, Relyvrio, to be “very promising,” which is why he testified to the FDA in favor of approval. (He’s a consultant and disease state speaker for multiple companies including Amylyx, manufacturer of Relyvrio.)
The “drug has different mechanisms of action than the currently approved treatments,” Bedlack said. He added that, when Relyvrio is taken in addition to Riluzole, it appears to slow functional decline by an additional 25 percent and extend life by another 6 to 10 months. “It is not a cure, but it is definitely a step forward.”
T. Scott Diesing, a neurohospitalist and director of general neurology at the University of Nebraska Medical Center in Omaha, said he hopes the drug is “as good as people anticipated it should be, because there are not too many options for these patients.”
"FDA went out on a limb in approving Relyvrio based on limited results from a small trial while a larger study remains in progress," said Florian P. Thomas, co-director of the ALS Center at Hackensack University Medical Center and Hackensack Meridian School of Medicine in New Jersey. "While it is definitely promising, clearly, the last word on this drug has not been spoken."
So far, Rouse's voice is holding up, but he knows the day will come when ALS will steal that and much more from him.
ALS is 100 percent fatal, with some patients dying as soon as a year after diagnosis. A few have lasted as long as 15 years, but those are the exceptions, Diesing said.
“If this drug can provide even months of additional life, or would maintain quality of life, that’s a big deal,” he noted, adding that “the patients are saying, ‘I know it’s not proven conclusively, but what do we have to lose?’ So, they would like to try it while additional studies are ongoing.” The drug has already been conditionally approved in Canada.
As his disease progresses, Rouse hopes to get a speech-to-text voice-generating computer that he can control with his eyes. So far, his voice is holding up, but he knows the day will come when ALS will steal that and much more from him. He works at I AM ALS, a patient-led community, and six of his friends have already died of the disease.
“Every time I lose a friend to ALS, I grieve and am sad but I resolve myself to keep working harder for them, myself and others,” Rouse said. “People living with ALS find great purpose in life advocating and trying to make a difference.”
Friday Five Podcast: New drug may slow the rate of Alzheimer's disease
On September 27, pharmaceuticals Biogen and Eisai announced that their drug, lecanemab, can slow the rate of Alzheimer's disease, according to a clinical trial. Today's Friday Five episode covers this story and other health research over the month of September.
The Friday Five covers important stories in health and science research that you may have missed - usually over the previous week, but today's episode is a lookback on important studies over the month of September.
Most recently, on September 27, pharmaceuticals Biogen and Eisai announced that a clinical trial showed their drug, lecanemab, can slow the rate of Alzheimer's disease. There are plenty of controversies and troubling ethical issues in science – and we get into many of them in our online magazine – but this news roundup focuses on scientific creativity and progress to give you a therapeutic dose of inspiration headed into the weekend and the new month.
Listen on Apple | Listen on Spotify | Listen on Stitcher | Listen on Amazon | Listen on Google
This Friday Five episode covers the following studies published and announced over the past month:
- A new drug is shown to slow the rate of Alzheimer's disease
- The need for speed if you want to reduce your risk of dementia
- How to refreeze the north and south poles
- Ancient wisdom about Neti pots could pay off for Covid
- Two women, one man and a baby