
Sammy Basso is one of around 400 young people in the world with progeria.
Just 400 or so people in the world live with progeria: The mutation that causes it usually arises de novo, or "of new," meaning that it is not inherited but happens spontaneously during gestation. The challenge, as with all rare diseases, is that few cases means few treatments.
"When we first started, there was absolutely nothing out there," says Leslie Gordon, a physician-researcher who co-founded the Progeria Research Foundation in 1999 after her own son, also named Sam, was diagnosed with the disease. "We knew we had to jumpstart the entire field, so we collected money through road races and special events and writing grants and all sorts of donors… I think the first year we raised $75,000, most of it from one donor."
"We have not only the possibility but the responsibility to make the world a better world, and also to make a body a better body."
By 2003, the foundation had collaborated with Francis Collins, a geneticist who is now director of the National Institutes of Health, to work out the genetic basis for progeria—that single mutation Sammy has. The discovery led to interest in lonafarnib, a drug that was already being used in cancer patients but could potentially operate downstream of the mutation, preventing the buildup of the defective progerin in the body. "We funded cellular studies to look at a lonafarnib in cells, mouse studies to look at lonafarnib in mouse models of progeria… and then we initiated the clinical trials," Gordon says.
Sammy Basso's family had gotten involved with the Progeria Research Foundation through their international patient registry, which maintains relationships with families in 49 countries. "We started to hear about lonafarnib in 2006 from Leslie Gordon," says Sammy's father, Amerigo Basso, with his son translating. "She told us about the lonafarnib. And we were very happy because for the first time we understood that there was something that could help our son and our lives." Amerigo used the Italian word speranza, which means hope.
Still, Sammy wasn't sure if lonafarnib was right for him. "Since when I was very young I thought that everything happens for a reason. So, in my mind, if God made me with progeria, there was a reason, and to try to heal from progeria was something wrong," he says. Gradually, his parents and doctors, and Leslie Gordon, convinced him otherwise. Sammy began to believe that God was also the force behind doctors, science, and research. "And so we have not only the possibility but the responsibility to make the world a better world, and also to make a body a better body," he says.

Sammy Basso and his parents.
Courtesy of Basso
Sammy began taking lonafarnib, with the Progeria Research Foundation intermittently flying him, and other international trial participants, to Boston for tests. He was immediately beset by some of the drug's more unpleasant side effects: Stomach problems, nausea, and vomiting. "The first period was absolutely the worst period of my life," he says.
At first, doctors prescribed other medicines for the side effects, but to Sammy it had as much effect as drinking water. He visited doctor after doctor, with some calling him weekly or even daily to ask how he was doing. Eventually the specialists decided that he should lower his dose, balancing his pain with the benefit of the drug. Sammy can't actually feel any positive effect of the lonafarnib, but his health measurements have improved relative to people with progeria who don't take it.
While they never completely disappeared, Sammy's side effects decreased to the point that he could live. Inspired by the research that led to lonafarnib, he went to university to study molecular biology. For his thesis work, he travelled to Spain to perform experiments on cells and on mice with progeria, learning how to use the gene-editing technique CRISPR-Cas9 to cut out the mutated bit of DNA. "I was so excited to participate in this study," Sammy says. He felt like his work could make a difference.
In 2018, the Progeria Research Foundation was hosting one of their biennial workshops when Francis Collins, the researcher who had located the mutation behind progeria 15 years earlier, got in touch with Leslie Gordon. "Francis called me and said, Hey, I just saw a talk by David Liu from the Broad [Institute]. And it was pretty amazing. He has been looking at progeria and has very early, but very exciting data… Do you have any spaces, any slots you could make in your program for late breaking news?"
Gordon found a spot, and David Liu came to talk about what was going on in his lab, which was an even more advanced treatment that led to mice with the progeria mutation living into their senior mouse years—substantially closer to a normal lifespan. Liu's lab had built on the idea of CRISPR-Cas9 to create a more elegant genetic process called base editing: Instead of chopping out mutated DNA, a scientist could chemically convert an incorrect DNA letter to the correct one, like the search and replace function in word processing software. Mice who had their Lamin-A mutations corrected this way lived more than twice as long as untreated animals.
Sammy was in the audience at Dr. Liu's talk. "When I heard about this base editing as a younger scientist, I thought that I was living in the future," he says. "When my parents had my diagnosis of progeria, the science knew very little information about DNA. And now we are talking about healing the DNA… It is incredible."
Lonafarnib (also called Zokinvy) was approved by the US Food and Drug Administration this past November. Sammy, now 25, still takes it, and still manages his side effects. With luck, the gift of a few extra years will act as a bridge until he can try Liu's revolutionary new gene treatment, which has not yet begun testing in humans. While Leslie Gordon warns that she's always wrong about things like this, she hopes to see the new base editing techniques in clinical trials in the next year or two. Sammy won't need to be convinced to try it this time; his thinking on fate has evolved since his first encounter with lonafarnib.
"I would be very happy to try it," he says. "I know that for a non-scientist it can be difficult to understand. Some people think that we are the DNA. We are not. The DNA is a part of us, and to correct it is to do what we are already doing—just better." In short, a gene therapy, while it may seem like science fiction, is no different from a pill. For Sammy, both are a new way to think about fate: No longer something that simply happens to him.

Thanks to safety cautions from the COVID-19 pandemic, a strain of influenza has been completely eliminated.
The flu shot, explained
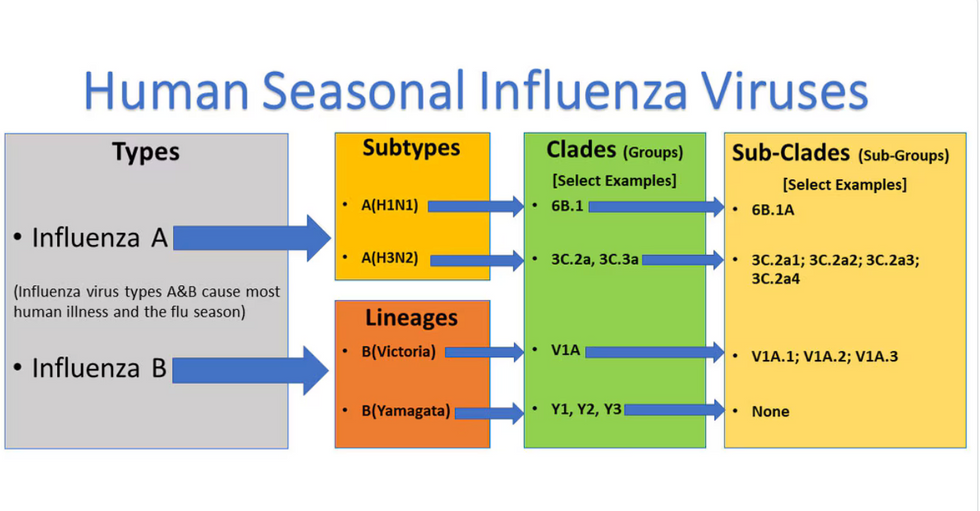
Influenza viruses type A and B are responsible for the majority of human illnesses and the flu season.
Centers for Disease Control
For more than a decade, flu shots have protected against two types of the influenza virus–type A and type B. While there are four different strains of influenza in existence (A, B, C, and D), only strains A, B, and C are capable of infecting humans, and only A and B cause pandemics. In other words, if you catch the flu during flu season, you’re most likely sick with flu type A or B.
Flu vaccines contain inactivated—or dead—influenza virus. These inactivated viruses can’t cause sickness in humans, but when administered as part of a vaccine, they teach a person’s immune system to recognize and kill those viruses when they’re encountered in the wild.
Each spring, a panel of experts gives a recommendation to the US Food and Drug Administration on which strains of each flu type to include in that year’s flu vaccine, depending on what surveillance data says is circulating and what they believe is likely to cause the most illness during the upcoming flu season. For the past decade, Americans have had access to vaccines that provide protection against two strains of influenza A and two lineages of influenza B, known as the Victoria lineage and the Yamagata lineage. But this year, the seasonal flu shot won’t include the Yamagata strain, because the Yamagata strain is no longer circulating among humans.
How Yamagata Disappeared

Flu surveillance data from the Global Initiative on Sharing All Influenza Data (GISAID) shows that the Yamagata lineage of flu type B has not been sequenced since April 2020.
Nature
Experts believe that the Yamagata lineage had already been in decline before the pandemic hit, likely because the strain was naturally less capable of infecting large numbers of people compared to the other strains. When the COVID-19 pandemic hit, the resulting safety precautions such as social distancing, isolating, hand-washing, and masking were enough to drive the virus into extinction completely.
Because the strain hasn’t been circulating since 2020, the FDA elected to remove the Yamagata strain from the seasonal flu vaccine. This will mark the first time since 2012 that the annual flu shot will be trivalent (three-component) rather than quadrivalent (four-component).
Should I still get the flu shot?
The flu shot will protect against fewer strains this year—but that doesn’t mean we should skip it. Influenza places a substantial health burden on the United States every year, responsible for hundreds of thousands of hospitalizations and tens of thousands of deaths. The flu shot has been shown to prevent millions of illnesses each year (more than six million during the 2022-2023 season). And while it’s still possible to catch the flu after getting the flu shot, studies show that people are far less likely to be hospitalized or die when they’re vaccinated.
Another unexpected benefit of dropping the Yamagata strain from the seasonal vaccine? This will possibly make production of the flu vaccine faster, and enable manufacturers to make more vaccines, helping countries who have a flu vaccine shortage and potentially saving millions more lives.

Founder Lewis Hornby and his grandmother Pat, sampling Jelly Drops—an edible gummy containing water and life-saving electrolytes.
When Lewis Hornby visited his grandmother at her nursing home afterward, he learned that dehydration especially affects people with dementia, as they often don’t feel thirst cues at all, or may not recognize how to use cups correctly. But while dementia patients often don’t remember to drink water, it seemed to Hornby that they had less problem remembering to eat, particularly candy.
 Where people with dementia often forget to drink water, they're more likely to pick up a colorful snack, Hornby found. alzheimers.org.uk
Where people with dementia often forget to drink water, they're more likely to pick up a colorful snack, Hornby found. alzheimers.org.uk
Hornby wanted to create a solution for elderly people who struggled keeping their fluid intake up. He spent the next eighteen months researching and designing a solution and securing funding for his project. In 2019, Hornby won a sizable grant from the Alzheimer’s Society, a UK-based care and research charity for people with dementia and their caregivers. Together, through the charity’s Accelerator Program, they created a bite-sized, sugar-free, edible jelly drop that looked and tasted like candy. The candy, called Jelly Drops, contained 95% water and electrolytes—important minerals that are often lost during dehydration. The final product launched in 2020—and was an immediate success. The drops were able to provide extra hydration to the elderly, as well as help keep dementia patients safe, since dehydration commonly leads to confusion, hospitalization, and sometimes even death.
Not only did Jelly Drops quickly become a favorite snack among dementia patients in the UK, but they were able to provide an additional boost of hydration to hospital workers during the pandemic. In NHS coronavirus hospital wards, patients infected with the virus were regularly given Jelly Drops to keep their fluid levels normal—and staff members snacked on them as well, since long shifts and personal protective equipment (PPE) they were required to wear often left them feeling parched.
In April 2022, Jelly Drops launched in the United States. The company continues to donate 1% of its profits to help fund Alzheimer’s research.