New research focuses on methods that could change medicine-making worldwide. The scientists propose bursting cells open, removing their DNA and using the cellular gears inside to make therapies.
Rao and his team of collaborators, which spans multiple research institutions, believe they have a better approach that may change medicine-making worldwide. They suggest forgoing the concept of using living cells as medicine-producers. Instead, they propose breaking the cells and using the remaining cellular gears for assembling the therapeutic compounds. Instead of inserting the DNA into living cells, the team burst them open, and removed their DNA altogether. Yet, the residual molecular machinery of ribosomes, polymerases and other cogwheels still functioned the way it would in a cell. “Now if you drop your DNA drug-making instructions into that soup, this machinery starts making what you need,” Rao explains. “And because you're no longer worrying about living cells, it becomes much simpler and more efficient.” The collaborators detail their cell-free protein synthesis or CFPS method in their recent paper published in preprint BioAxiv.
While CFPS does not use living cells, it still needs the basic building blocks to assemble proteins from—such as amino acids, nucleotides and certain types of enzymes. These are regularly added into this “soup” to keep the molecular factory chugging. “We just mix everything in as a batch and we let it integrate,” says James Robert Swartz, professor of chemical engineering and bioengineering at Stanford University and co-author of the paper. “And we make sure that we provide enough oxygen.” Rao likens the process to making milk from milk powder.
For a variety of reasons—from the field’s general inertia to regulatory approval hurdles—the method hasn’t become mainstream. The pandemic rekindled interest in medicines that can be made quickly and easily, so it drew more attention to the technology.
The idea of a cell-free protein synthesis is older than one might think. Swartz first experimented with it around 1997, when he was a chemical engineer at Genentech. While working on engineering bacteria to make pharmaceuticals, he discovered that there was a limit to what E. coli cells, the workhorse darling of pharma, could do. For example, it couldn’t grow and properly fold some complex proteins. “We tried many genetic engineering approaches, many fermentation, development, and environmental control approaches,” Swartz recalls—to no avail.
“The organism had its own agenda,” he quips. “And because everything was happening within the organism, we just couldn't really change those conditions very easily. Some of them we couldn’t change at all—we didn’t have control.”
It was out of frustration with the defiant bacteria that a new idea took hold. Could the cells be opened instead, so that the protein-forming reactions could be influenced more easily? “Obviously, we’d lose the ability for them to reproduce,” Swartz says. But that also meant that they no longer needed to keep the cells alive and could focus on making the specific reactions happen. “We could take the catalysts, the enzymes, and the more complex catalysts and activate them, make them work together, much as they would in a living cell, but the way we wanted.”
In 1998, Swartz joined Stanford, and began perfecting the biochemistry of the cell-free method, identifying the reactions he wanted to foster and stopping those he didn’t want. He managed to make the idea work, but for a variety of reasons—from the field’s general inertia to regulatory approval hurdles—the method hasn’t become mainstream. The pandemic rekindled interest in medicines that can be made quickly and easily, so it drew more attention to the technology. For their BioArxiv paper, the team tested the method by growing a specific antiviral protein called griffithsin.
First identified by Barry O’Keefe at National Cancer Institute over a decade ago, griffithsin is an antiviral known to interfere with many viruses’ ability to enter cells—including HIV, SARS, SARS-CoV-2, MERS and others. Originally isolated from the red algae Griffithsia, it works differently from antibodies and antibody cocktails.
Most antiviral medicines tend to target the specific receptors that viruses use to gain entry to the cells they infect. For example, SARS-CoV-2 uses the infamous spike protein to latch onto the ACE2 receptor of mammalian cells. The antibodies or other antiviral molecules stick to the spike protein, shutting off its ability to cling onto the ACE2 receptors. Unfortunately, the spike proteins mutate very often, so the medicines lose their potency. On the contrary, griffithsin has the ability to cling to the different parts of viral shells called capsids—namely to the molecules of mannose, a type of sugar. That extra stuff, glued all around the capsid like dead weight, makes it impossible for the virus to squeeze into the cell.
“Every time we have a vaccine or an antibody against a specific SARS-CoV-2 strain, that strain then mutates and so you lose efficacy,” Rao explains. “But griffithsin molecules glom onto the viral capsid, so the capsid essentially becomes a sticky mess and can’t enter the cell.” Mannose molecules also don’t mutate as easily as viruses’ receptors, so griffithsin-based antivirals do not have to be constantly updated. And because mannose molecules are found on many viruses’ capsids, it makes griffithsin “a universal neutralizer,” Rao explains.
“When griffithsin was discovered, we recognized that it held a lot of promise as a potential antiviral agent,” O’Keefe says. In 2010, he published a paper about griffithsin efficacy in neutralizing viruses of the corona family—after the first SARS outbreak in the early 2000s, the scientific community was interested in such antivirals. Yet, griffithsin is still not available as an off-the-shelf product. So during the Covid pandemic, the team experimented with synthesizing griffithsin using the cell-free production method. They were able to generate potent griffithsin in less than 24 hours without having to grow living cells.
The antiviral protein isn't the only type of medicine that can be made cell-free. The proteins needed for vaccine production could also be made the same way. “Such portable, on-demand drug manufacturing platforms can produce antiviral proteins within hours, making them ideal for combating future pandemics,” Rao says. “We would be able to stop the pandemic before it spreads.”
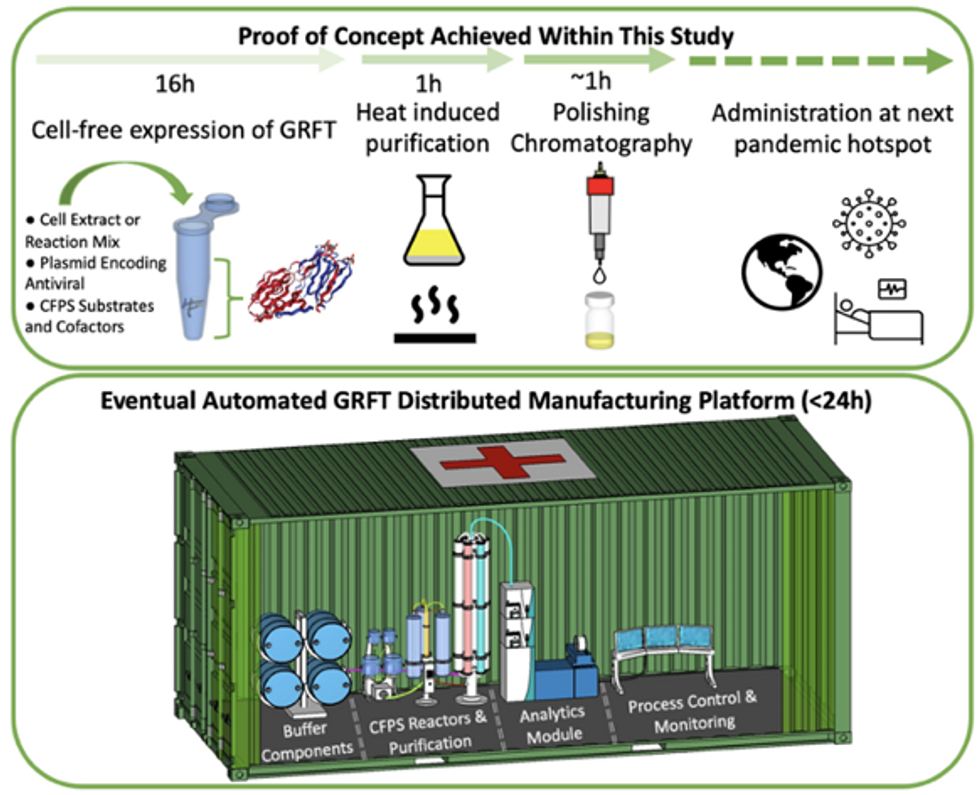
Top: Describes the process used in the study. Bottom: Describes how the new medicines and vaccines could be made at the site of a future viral outbreak.
Image courtesy of Rao and team, sourced from An approach to rapid distributed manufacturing of broad spectrumanti-viral griffithsin using cell-free systems to mitigate pandemics.
Rao’s idea is to perfect the technology to the point that any hospital or pharmacy can load up the media containing molecular factories, mix up the required amino acids, nucleotides and enzymes, and harvest the meds within hours. That will allow making medicines onsite and on demand. “That would be a self-contained production unit, so that you could just ship the production wherever the pandemic is breaking out,” says Swartz.
These units and the meds they produce, will, of course, have to undergo rigorous testing. “The biggest hurdles will be validating these against conventional technology,” Rao says. The biotech industry is risk-averse and prefers the familiar methods. But if this approach works, it may go beyond emergency situations and revolutionize the medicine-making paradigm even outside hospitals and pharmacies. Rao hopes that someday the method might become so mainstream that people may be able to buy and operate such reactors at home. “You can imagine a diabetic patient making insulin that way, or some other drugs,” Rao says. It would work not unlike making baby formula from the mere white powder. Just add water—and some oxygen, too.