

A newborn and mother in the hospital - first touch. (© martin81/Shutterstock)
Today in Melrose, Massachusetts, Cora Stetson is the picture of good health, a bubbly precocious 2-year-old. But Cora has two separate mutations in the gene that produces a critical enzyme called biotinidase and her body produces only 40 percent of the normal levels of that enzyme.
In the last few years, the dream of predicting and preventing diseases through genomics, starting in childhood, is finally within reach.
That's enough to pass conventional newborn (heelstick) screening, but may not be enough for normal brain development, putting baby Cora at risk for seizures and cognitive impairment. But thanks to an experimental study in which Cora's DNA was sequenced after birth, this condition was discovered and she is being treated with a safe and inexpensive vitamin supplement.
Stories like these are beginning to emerge from the BabySeq Project, the first clinical trial in the world to systematically sequence healthy newborn infants. This trial was led by my research group with funding from the National Institutes of Health. While still controversial, it is pointing the way to a future in which adults, or even newborns, can receive comprehensive genetic analysis in order to determine their risk of future disease and enable opportunities to prevent them.
Some believe that medicine is still not ready for genomic population screening, but others feel it is long overdue. After all, the sequencing of the Human Genome Project was completed in 2003, and with this milestone, it became feasible to sequence and interpret the genome of any human being. The costs have come down dramatically since then; an entire human genome can now be sequenced for about $800, although the costs of bioinformatic and medical interpretation can add another $200 to $2000 more, depending upon the number of genes interrogated and the sophistication of the interpretive effort.

Two-year-old Cora Stetson, whose DNA sequencing after birth identified a potentially dangerous genetic mutation in time for her to receive preventive treatment.
(Photo courtesy of Robert Green)
The ability to sequence the human genome yielded extraordinary benefits in scientific discovery, disease diagnosis, and targeted cancer treatment. But the ability of genomes to detect health risks in advance, to actually predict the medical future of an individual, has been mired in controversy and slow to manifest. In particular, the oft-cited vision that healthy infants could be genetically tested at birth in order to predict and prevent the diseases they would encounter, has proven to be far tougher to implement than anyone anticipated.
But in the last few years, the dream of predicting and preventing diseases through genomics, starting in childhood, is finally within reach. Why did it take so long? And what remains to be done?
Great Expectations
Part of the problem was the unrealistic expectations that had been building for years in advance of the genomic science itself. For example, the 1997 film Gattaca portrayed a near future in which the lifetime risk of disease was readily predicted the moment an infant is born. In the fanfare that accompanied the completion of the Human Genome Project, the notion of predicting and preventing future disease in an individual became a powerful meme that was used to inspire investment and public support for genomic research long before the tools were in place to make it happen.
Another part of the problem was the success of state-mandated newborn screening programs that began in the 1960's with biochemical tests of the "heel-stick" for babies with metabolic disorders. These programs have worked beautifully, costing only a few dollars per baby and saving thousands of infants from death and severe cognitive impairment. It seemed only logical that a new technology like genome sequencing would add power and promise to such programs. But instead of embracing the notion of newborn sequencing, newborn screening laboratories have thus far rejected the entire idea as too expensive, too ambiguous, and too threatening to the comfortable constituency that they had built within the public health framework.
"What can you find when you look as deeply as possible into the medical genomes of healthy individuals?"
Creating the Evidence Base for Preventive Genomics
Despite a number of obstacles, there are researchers who are exploring how to achieve the original vision of genomic testing as a tool for disease prediction and prevention. For example, in our NIH-funded MedSeq Project, we were the first to ask the question: "What can you find when you look as deeply as possible into the medical genomes of healthy individuals?"
Most people do not understand that genetic information comes in four separate categories: 1) dominant mutations putting the individual at risk for rare conditions like familial forms of heart disease or cancer, (2) recessive mutations putting the individual's children at risk for rare conditions like cystic fibrosis or PKU, (3) variants across the genome that can be tallied to construct polygenic risk scores for common conditions like heart disease or type 2 diabetes, and (4) variants that can influence drug metabolism or predict drug side effects such as the muscle pain that occasionally occurs with statin use.
The technological and analytical challenges of our study were formidable, because we decided to systematically interrogate over 5000 disease-associated genes and report results in all four categories of genetic information directly to the primary care physicians for each of our volunteers. We enrolled 200 adults and found that everyone who was sequenced had medically relevant polygenic and pharmacogenomic results, over 90 percent carried recessive mutations that could have been important to reproduction, and an extraordinary 14.5 percent carried dominant mutations for rare genetic conditions.
A few years later we launched the BabySeq Project. In this study, we restricted the number of genes to include only those with child/adolescent onset that could benefit medically from early warning, and even so, we found 9.4 percent carried dominant mutations for rare conditions.
At first, our interpretation around the high proportion of apparently healthy individuals with dominant mutations for rare genetic conditions was simple – that these conditions had lower "penetrance" than anticipated; in other words, only a small proportion of those who carried the dominant mutation would get the disease. If this interpretation were to hold, then genetic risk information might be far less useful than we had hoped.
Suddenly the information available in the genome of even an apparently healthy individual is looking more robust, and the prospect of preventive genomics is looking feasible.
But then we circled back with each adult or infant in order to examine and test them for any possible features of the rare disease in question. When we did this, we were surprised to see that in over a quarter of those carrying such mutations, there were already subtle signs of the disease in question that had not even been suspected! Now our interpretation was different. We now believe that genetic risk may be responsible for subclinical disease in a much higher proportion of people than has ever been suspected!
Meanwhile, colleagues of ours have been demonstrating that detailed analysis of polygenic risk scores can identify individuals at high risk for common conditions like heart disease. So adding up the medically relevant results in any given genome, we start to see that you can learn your risks for a rare monogenic condition, a common polygenic condition, a bad effect from a drug you might take in the future, or for having a child with a devastating recessive condition. Suddenly the information available in the genome of even an apparently healthy individual is looking more robust, and the prospect of preventive genomics is looking feasible.
Preventive Genomics Arrives in Clinical Medicine
There is still considerable evidence to gather before we can recommend genomic screening for the entire population. For example, it is important to make sure that families who learn about such risks do not suffer harms or waste resources from excessive medical attention. And many doctors don't yet have guidance on how to use such information with their patients. But our research is convincing many people that preventive genomics is coming and that it will save lives.
In fact, we recently launched a Preventive Genomics Clinic at Brigham and Women's Hospital where information-seeking adults can obtain predictive genomic testing with the highest quality interpretation and medical context, and be coached over time in light of their disease risks toward a healthier outcome. Insurance doesn't yet cover such testing, so patients must pay out of pocket for now, but they can choose from a menu of genetic screening tests, all of which are more comprehensive than consumer-facing products. Genetic counseling is available but optional. So far, this service is for adults only, but sequencing for children will surely follow soon.
As the costs of sequencing and other Omics technologies continue to decline, we will see both responsible and irresponsible marketing of genetic testing, and we will need to guard against unscientific claims. But at the same time, we must be far more imaginative and fast moving in mainstream medicine than we have been to date in order to claim the emerging benefits of preventive genomics where it is now clear that suffering can be averted, and lives can be saved. The future has arrived if we are bold enough to grasp it.
Funding and Disclosures:
Dr. Green's research is supported by the National Institutes of Health, the Department of Defense and through donations to The Franca Sozzani Fund for Preventive Genomics. Dr. Green receives compensation for advising the following companies: AIA, Applied Therapeutics, Helix, Ohana, OptraHealth, Prudential, Verily and Veritas; and is co-founder and advisor to Genome Medical, Inc, a technology and services company providing genetics expertise to patients, providers, employers and care systems.

Recent advancements in engineering mean that the first preclinical trials for an artificial kidney could happen soon.
Still, the devices aren’t ready for testing in humans—yet. But recent advancements in engineering mean that the first preclinical trials for an artificial kidney could happen soon, according to Jonathan Himmelfarb, a nephrologist at the University of Washington.
“It would liberate people with kidney failure,” Himmelfarb says.
An engineering marvel
Compared to the heart or the brain, the kidney doesn’t get as much respect from the medical profession, but its job is far more complex. “It does hundreds of different things,” says UCLA’s Ira Kurtz.
Kurtz would know. He’s worked as a nephrologist for 37 years, devoting his career to helping those with kidney disease. While his colleagues in cardiology and endocrinology have seen major advances in the development of artificial hearts and insulin pumps, little has changed for patients on hemodialysis. The machines remain bulky and require large volumes of a liquid called dialysate to remove toxins from a patient’s blood, along with gallons of purified water. A kidney transplant is the next best thing to someone’s own, functioning organ, but with over 600,000 Americans on dialysis and only about 100,000 kidney transplants each year, most of those in kidney failure are stuck on dialysis.
Part of the lack of progress in artificial kidney design is the sheer complexity of the kidney’s job. Each of the 45 different cell types in the kidney do something different.
Part of the lack of progress in artificial kidney design is the sheer complexity of the kidney’s job. To build an artificial heart, Kurtz says, you basically need to engineer a pump. An artificial pancreas needs to balance blood sugar levels with insulin secretion. While neither of these tasks is simple, they are fairly straightforward. The kidney, on the other hand, does more than get rid of waste products like urea and other toxins. Each of the 45 different cell types in the kidney do something different, helping to regulate electrolytes like sodium, potassium, and phosphorous; maintaining blood pressure and water balance; guiding the body’s hormonal and inflammatory responses; and aiding in the formation of red blood cells.

There's been little progress for patients during Ira Kurtz's 37 years as a nephrologist. Artificial kidneys would change that.
UCLA
Dialysis primarily filters waste, and does so well enough to keep someone alive, but it isn’t a true artificial kidney because it doesn’t perform the kidney’s other jobs, according to Kurtz, such as sensing levels of toxins, wastes, and electrolytes in the blood. Due to the size and water requirements of existing dialysis machines, the equipment isn’t portable. Physicians write a prescription for a certain duration of dialysis and assess how well it’s working with semi-regular blood tests. The process of dialysis itself, however, is conducted blind. Doctors can’t tell how much dialysis a patient needs based on kidney values at the time of treatment, says Meera Harhay, a nephrologist at Drexel University in Philadelphia.
But it’s the impact of dialysis on their day-to-day lives that creates the most problems for patients. Only one-quarter of those on dialysis are able to remain employed (compared to 85% of similar-aged adults), and many report a low quality of life. Having more flexibility in life would make a major different to her patients, Harhay says.
“Almost half their week is taken up by the burden of their treatment. It really eats away at their freedom and their ability to do things that add value to their life,” she says.
Art imitates life
The challenge for artificial kidney designers was how to compress the kidney’s natural functions into a portable, wearable, or implantable device that wouldn’t need constant access to gallons of purified and sterilized water. The other universal challenge they faced was ensuring that any part of the artificial kidney that would come in contact with blood was kept germ-free to prevent infection.
As part of the 2021 KidneyX Prize, a partnership between the U.S. Department of Health and Human Services and the American Society of Nephrology, inventors were challenged to create prototypes for artificial kidneys. Himmelfarb’s team at the University of Washington’s Center for Dialysis Innovation won the prize by focusing on miniaturizing existing technologies to create a portable dialysis machine. The backpack sized AKTIV device (Ambulatory Kidney to Increase Vitality) will recycle dialysate in a closed loop system that removes urea from blood and uses light-based chemical reactions to convert the urea to nitrogen and carbon dioxide, which allows the dialysate to be recirculated.
Himmelfarb says that the AKTIV can be used when at home, work, or traveling, which will give users more flexibility and freedom. “If you had a 30-pound device that you could put in the overhead bins when traveling, you could go visit your grandkids,” he says.
Kurtz’s team at UCLA partnered with the U.S. Kidney Research Corporation and Arkansas University to develop a dialysate-free desktop device (about the size of a small printer) as the first phase of a progression that will he hopes will lead to something small and implantable. Part of the reason for the artificial kidney’s size, Kurtz says, is the number of functions his team are cramming into it. Not only will it filter urea from blood, but it will also use electricity to help regulate electrolyte levels in a process called electrodeionization. Kurtz emphasizes that these additional functions are what makes his design a true artificial kidney instead of just a small dialysis machine.

One version of an artificial kidney.
UCLA
“It doesn't have just a static function. It has a bank of sensors that measure chemicals in the blood and feeds that information back to the device,” Kurtz says.
Other startups are getting in on the game. Nephria Bio, a spinout from the South Korean-based EOFlow, is working to develop a wearable dialysis device, akin to an insulin pump, that uses miniature cartridges with nanomaterial filters to clean blood (Harhay is a scientific advisor to Nephria). Ian Welsford, Nephria’s co-founder and CTO, says that the device’s design means that it can also be used to treat acute kidney injuries in resource-limited settings. These potentials have garnered interest and investment in artificial kidneys from the U.S. Department of Defense.
For his part, Burton is most interested in an implantable device, as that would give him the most freedom. Even having a regular outpatient procedure to change batteries or filters would be a minor inconvenience to him.
“Being plugged into a machine, that’s not mimicking life,” he says.
This article was first published by Leaps.org on May 5, 2022.

New research focuses on methods that could change medicine-making worldwide. The scientists propose bursting cells open, removing their DNA and using the cellular gears inside to make therapies.
Rao and his team of collaborators, which spans multiple research institutions, believe they have a better approach that may change medicine-making worldwide. They suggest forgoing the concept of using living cells as medicine-producers. Instead, they propose breaking the cells and using the remaining cellular gears for assembling the therapeutic compounds. Instead of inserting the DNA into living cells, the team burst them open, and removed their DNA altogether. Yet, the residual molecular machinery of ribosomes, polymerases and other cogwheels still functioned the way it would in a cell. “Now if you drop your DNA drug-making instructions into that soup, this machinery starts making what you need,” Rao explains. “And because you're no longer worrying about living cells, it becomes much simpler and more efficient.” The collaborators detail their cell-free protein synthesis or CFPS method in their recent paper published in preprint BioAxiv.
While CFPS does not use living cells, it still needs the basic building blocks to assemble proteins from—such as amino acids, nucleotides and certain types of enzymes. These are regularly added into this “soup” to keep the molecular factory chugging. “We just mix everything in as a batch and we let it integrate,” says James Robert Swartz, professor of chemical engineering and bioengineering at Stanford University and co-author of the paper. “And we make sure that we provide enough oxygen.” Rao likens the process to making milk from milk powder.
For a variety of reasons—from the field’s general inertia to regulatory approval hurdles—the method hasn’t become mainstream. The pandemic rekindled interest in medicines that can be made quickly and easily, so it drew more attention to the technology.
The idea of a cell-free protein synthesis is older than one might think. Swartz first experimented with it around 1997, when he was a chemical engineer at Genentech. While working on engineering bacteria to make pharmaceuticals, he discovered that there was a limit to what E. coli cells, the workhorse darling of pharma, could do. For example, it couldn’t grow and properly fold some complex proteins. “We tried many genetic engineering approaches, many fermentation, development, and environmental control approaches,” Swartz recalls—to no avail.
“The organism had its own agenda,” he quips. “And because everything was happening within the organism, we just couldn't really change those conditions very easily. Some of them we couldn’t change at all—we didn’t have control.”
It was out of frustration with the defiant bacteria that a new idea took hold. Could the cells be opened instead, so that the protein-forming reactions could be influenced more easily? “Obviously, we’d lose the ability for them to reproduce,” Swartz says. But that also meant that they no longer needed to keep the cells alive and could focus on making the specific reactions happen. “We could take the catalysts, the enzymes, and the more complex catalysts and activate them, make them work together, much as they would in a living cell, but the way we wanted.”
In 1998, Swartz joined Stanford, and began perfecting the biochemistry of the cell-free method, identifying the reactions he wanted to foster and stopping those he didn’t want. He managed to make the idea work, but for a variety of reasons—from the field’s general inertia to regulatory approval hurdles—the method hasn’t become mainstream. The pandemic rekindled interest in medicines that can be made quickly and easily, so it drew more attention to the technology. For their BioArxiv paper, the team tested the method by growing a specific antiviral protein called griffithsin.
First identified by Barry O’Keefe at National Cancer Institute over a decade ago, griffithsin is an antiviral known to interfere with many viruses’ ability to enter cells—including HIV, SARS, SARS-CoV-2, MERS and others. Originally isolated from the red algae Griffithsia, it works differently from antibodies and antibody cocktails.
Most antiviral medicines tend to target the specific receptors that viruses use to gain entry to the cells they infect. For example, SARS-CoV-2 uses the infamous spike protein to latch onto the ACE2 receptor of mammalian cells. The antibodies or other antiviral molecules stick to the spike protein, shutting off its ability to cling onto the ACE2 receptors. Unfortunately, the spike proteins mutate very often, so the medicines lose their potency. On the contrary, griffithsin has the ability to cling to the different parts of viral shells called capsids—namely to the molecules of mannose, a type of sugar. That extra stuff, glued all around the capsid like dead weight, makes it impossible for the virus to squeeze into the cell.
“Every time we have a vaccine or an antibody against a specific SARS-CoV-2 strain, that strain then mutates and so you lose efficacy,” Rao explains. “But griffithsin molecules glom onto the viral capsid, so the capsid essentially becomes a sticky mess and can’t enter the cell.” Mannose molecules also don’t mutate as easily as viruses’ receptors, so griffithsin-based antivirals do not have to be constantly updated. And because mannose molecules are found on many viruses’ capsids, it makes griffithsin “a universal neutralizer,” Rao explains.
“When griffithsin was discovered, we recognized that it held a lot of promise as a potential antiviral agent,” O’Keefe says. In 2010, he published a paper about griffithsin efficacy in neutralizing viruses of the corona family—after the first SARS outbreak in the early 2000s, the scientific community was interested in such antivirals. Yet, griffithsin is still not available as an off-the-shelf product. So during the Covid pandemic, the team experimented with synthesizing griffithsin using the cell-free production method. They were able to generate potent griffithsin in less than 24 hours without having to grow living cells.
The antiviral protein isn't the only type of medicine that can be made cell-free. The proteins needed for vaccine production could also be made the same way. “Such portable, on-demand drug manufacturing platforms can produce antiviral proteins within hours, making them ideal for combating future pandemics,” Rao says. “We would be able to stop the pandemic before it spreads.”
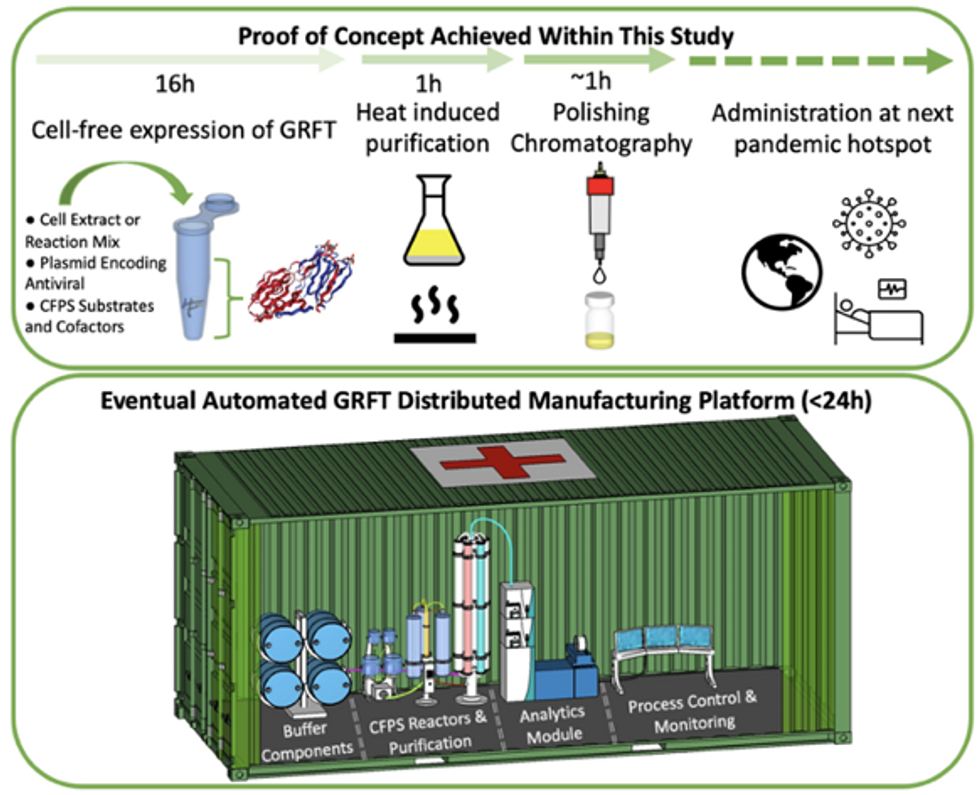
Top: Describes the process used in the study. Bottom: Describes how the new medicines and vaccines could be made at the site of a future viral outbreak.
Image courtesy of Rao and team, sourced from An approach to rapid distributed manufacturing of broad spectrumanti-viral griffithsin using cell-free systems to mitigate pandemics.
Rao’s idea is to perfect the technology to the point that any hospital or pharmacy can load up the media containing molecular factories, mix up the required amino acids, nucleotides and enzymes, and harvest the meds within hours. That will allow making medicines onsite and on demand. “That would be a self-contained production unit, so that you could just ship the production wherever the pandemic is breaking out,” says Swartz.
These units and the meds they produce, will, of course, have to undergo rigorous testing. “The biggest hurdles will be validating these against conventional technology,” Rao says. The biotech industry is risk-averse and prefers the familiar methods. But if this approach works, it may go beyond emergency situations and revolutionize the medicine-making paradigm even outside hospitals and pharmacies. Rao hopes that someday the method might become so mainstream that people may be able to buy and operate such reactors at home. “You can imagine a diabetic patient making insulin that way, or some other drugs,” Rao says. It would work not unlike making baby formula from the mere white powder. Just add water—and some oxygen, too.