
Is Finding Out Your Baby’s Genetics A New Responsibility of Parenting?
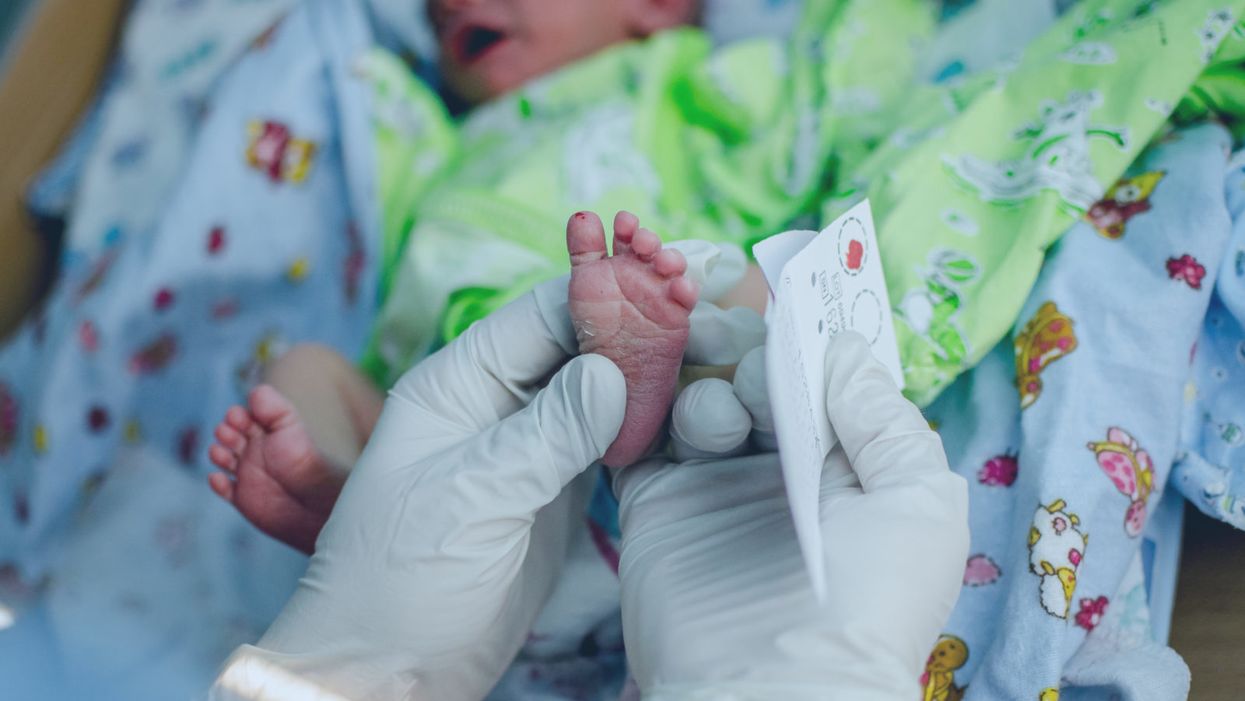
A doctor pricks the heel of a newborn for a blood test.
Hours after a baby is born, its heel is pricked with a lancet. Drops of the infant's blood are collected on a porous card, which is then mailed to a state laboratory. The dried blood spots are screened for around thirty conditions, including phenylketonuria (PKU), the metabolic disorder that kick-started this kind of newborn screening over 60 years ago. In the U.S., parents are not asked for permission to screen their child. Newborn screening programs are public health programs, and the assumption is that no good parent would refuse a screening test that could identify a serious yet treatable condition in their baby.
Learning as much as you can about your child's health might seem like a natural obligation of parenting. But it's an assumption that I think needs to be much more closely examined.
Today, with the introduction of genome sequencing into clinical medicine, some are asking whether newborn screening goes far enough. As the cost of sequencing falls, should parents take a more expansive look at their children's health, learning not just whether they have a rare but treatable childhood condition, but also whether they are at risk for untreatable conditions or for diseases that, if they occur at all, will strike only in adulthood? Should genome sequencing be a part of every newborn's care?
It's an idea that appeals to Anne Wojcicki, the founder and CEO of the direct-to-consumer genetic testing company 23andMe, who in a 2016 interview with The Guardian newspaper predicted that having newborns tested would soon be considered standard practice—"as critical as testing your cholesterol"—and a new responsibility of parenting. Wojcicki isn't the only one excited to see everyone's genes examined at birth. Francis Collins, director of the National Institutes of Health and perhaps the most prominent advocate of genomics in the United States, has written that he is "almost certain … that whole-genome sequencing will become part of new-born screening in the next few years." Whether that would happen through state-mandated screening programs, or as part of routine pediatric care—or perhaps as a direct-to-consumer service that parents purchase at birth or receive as a baby-shower gift—is not clear.
Learning as much as you can about your child's health might seem like a natural obligation of parenting. But it's an assumption that I think needs to be much more closely examined, both because the results that genome sequencing can return are more complex and more uncertain than one might expect, and because parents are not actually responsible for their child's lifelong health and well-being.
What is a parent supposed to do about such a risk except worry?
Existing newborn screening tests look for the presence of rare conditions that, if identified early in life, before the child shows any symptoms, can be effectively treated. Sequencing could identify many of these same kinds of conditions (and it might be a good tool if it could be targeted to those conditions alone), but it would also identify gene variants that confer an increased risk rather than a certainty of disease. Occasionally that increased risk will be significant. About 12 percent of women in the general population will develop breast cancer during their lives, while those who have a harmful BRCA1 or BRCA2 gene variant have around a 70 percent chance of developing the disease. But for many—perhaps most—conditions, the increased risk associated with a particular gene variant will be very small. Researchers have identified over 600 genes that appear to be associated with schizophrenia, for example, but any one of those confers only a tiny increase in risk for the disorder. What is a parent supposed to do about such a risk except worry?
Sequencing results are uncertain in other important ways as well. While we now have the ability to map the genome—to create a read-out of the pairs of genetic letters that make up a person's DNA—we are still learning what most of it means for a person's health and well-being. Researchers even have a name for gene variants they think might be associated with a disease or disorder, but for which they don't have enough evidence to be sure. They are called "variants of unknown (or uncertain) significance (VUS), and they pop up in most people's sequencing results. In cancer genetics, where much research has been done, about 1 in 5 gene variants are reclassified over time. Most are downgraded, which means that a good number of VUS are eventually designated benign.
While one parent might reasonably decide to learn about their child's risk for a condition about which nothing can be done medically, a different, yet still thoroughly reasonable, parent might prefer to remain ignorant so that they can enjoy the time before their child is afflicted.
Then there's the puzzle of what to do about results that show increased risk or even certainty for a condition that we have no idea how to prevent. Some genomics advocates argue that even if a result is not "medically actionable," it might have "personal utility" because it allows parents to plan for their child's future needs, to enroll them in research, or to connect with other families whose children carry the same genetic marker.
Finding a certain gene variant in one child might inform parents' decisions about whether to have another—and if they do, about whether to use reproductive technologies or prenatal testing to select against that variant in a future child. I have no doubt that for some parents these personal utility arguments are persuasive, but notice how far we've now strayed from the serious yet treatable conditions that motivated governments to set up newborn screening programs, and to mandate such testing for all.
Which brings me to the other problem with the call for sequencing newborn babies: the idea that even if it's not what the law requires, it's what good parents should do. That idea is very compelling when we're talking about sequencing results that show a serious threat to the child's health, especially when interventions are available to prevent or treat that condition. But as I have shown, many sequencing results are not of this type.
While one parent might reasonably decide to learn about their child's risk for a condition about which nothing can be done medically, a different, yet still thoroughly reasonable, parent might prefer to remain ignorant so that they can enjoy the time before their child is afflicted. This parent might decide that the worry—and the hypervigilence it could inspire in them—is not in their child's best interest, or indeed in their own. This parent might also think that it should be up to the child, when he or she is older, to decide whether to learn about his or her risk for adult-onset conditions, especially given that many adults at high familial risk for conditions like Alzheimer's or Huntington's disease choose never to be tested. This parent will value the child's future autonomy and right not to know more than they value the chance to prepare for a health risk that won't strike the child until 40 or 50 years in the future.
Parents are not obligated to learn about their children's risk for a condition that cannot be prevented, has a small risk of occurring, or that would appear only in adulthood.
Contemporary understandings of parenting are famously demanding. We are asked to do everything within our power to advance our children's health and well-being—to act always in our children's best interests. Against that backdrop, the need to sequence every newborn baby's genome might seem obvious. But we should be skeptical. Many sequencing results are complex and uncertain. Parents are not obligated to learn about their children's risk for a condition that cannot be prevented, has a small risk of occurring, or that would appear only in adulthood. To suggest otherwise is to stretch parental responsibilities beyond the realm of childhood and beyond factors that parents can control.
An Investigational Drug Offers Hope to Patients with a Disabling Neuromuscular Disease

Robert Thomas was a devoted runner, gym goer, and crew member on a sailing team in San Diego when, in his 40s, he noticed that his range of movement was becoming more limited.
He thought he was just getting older, but when he was hiking an uphill trail in Lake Tahoe, he kept tripping over rocks. "I'd never had this happen before," Robert says. "I knew something was wrong but didn't know what it was."
It wasn't until age 50 when he was diagnosed with Charcot-Marie-Tooth disease. The genetic disorder damages the peripheral nerves, which connect the brain and spinal cord to the rest of the body. This network of nerves is responsible for relaying information and signals about sensation, movement, and motor coordination. Over time, the disease causes debilitating muscle weakness and the loss of limb control.
Charcot-Marie-Tooth usually presents itself in childhood or in a person's teens, but in some patients, like Robert, onset can be later in life. Symptoms may include muscle cramping, tingling, or burning. Many patients also have high foot arches or hammer toes — toes that curl from the middle joint instead of pointing forward. Those affected often have difficulty walking and may lose sensation in their lower legs, feet, hands, or forearms. One of the most common rare diseases, it affects around 130,000 people in the United States and 2.8 million worldwide.
Like many people with Charcot-Marie-Tooth, or CMT, Robert wears corrective braces on his legs to help with walking. Now 61, he can't run or sail anymore because of the disease, but he still works out regularly and can hike occasionally. CMT also affects his grip, so he has to use special straps while doing some exercises.
For the past few years, Robert has been participating in a clinical trial for an investigational CMT drug. He takes the liquid formulation every morning and evening using an oral syringe. Scientists are following patients like Robert to learn if their symptoms stabilize or improve while on the drug. Dubbed PXT300, the drug was designed by French biopharmaceutical company Pharnext and is the farthest along in development for CMT. If approved, it would be the first drug for the disease.
Currently, there's no cure for CMT, only supportive treatments like pain medication. Some individuals receive physical and occupational therapy. A drug for CMT could be a game-changer for patients whose quality of life is severely affected by the disease.
Genetic Underpinnings
CMT arises from mutations in genes that are responsible for creating and maintaining the myelin sheath — the insulating layer around nerves. Pharnext's drug is meant to treat patients with CMT1A, the most common form of the disease, which represents about half of CMT cases. Around 5% of those with CMT1A become severely disabled and end up in wheelchairs. People with CMT1A have an extra copy of the gene PMP22, which makes a protein that's needed to maintain the myelin sheath around peripheral nerves.
Typically, an individual inherits one copy of PMP22 from each parent. But a person with CMT1A receives a copy of PMP22 from one parent and two copies from a parent with the disease. This extra copy of the gene results in excess protein production, which damages the cells responsible for preserving and regenerating the myelin sheath, called Schwann cells.
The myelin sheath helps ensure that a signal from the brain gets carried to nerves in the muscles so that a part of the body can carry out a particular action or movement. This sheath is like the insulation on an electrical cord and the action is like a light bulb. If the insulation is fine, the light bulb turns on. But if the insulation is frayed, the light will flicker.
"The same happens to these patients," says David Horn Solomon, CEO of Pharnext. "The signal to their muscle is weak and flickers." Over time, their muscles become weaker and thinner.
The PMP22 gene has proven difficult to target with a drug because it's located in a protected space — the Schwann cells that make up the insulation around nerves. "There's not an easy way to tamp it down," Solomon says.
Another company, Acceleron Pharma of Cambridge, Massachusetts, was developing an injectable CMT drug meant to increase the strength of leg muscles. But the company halted development last year after the experimental drug failed in a mid-stage trial. While the drug led to a statistically significant increase in muscle volume, it didn't translate to improvements in muscle function or quality of life for trial participants.
Made by Design
Pharnext's drug, PXT3003, is a combination of three existing drugs — baclofen, a muscle relaxant; naltrexone, a drug that decreases the desire for alcohol and opioids; and sorbitol, a type of sugar alcohol.
The company designed the drug using its artificial intelligence platform, which screened 20,000 existing drugs to predict combinations that could inhibit the PMP22 gene and thereby lower protein production. The AI system narrowed the search to several hundreds of combinations and Pharnext tested around 75 of them in the lab before landing on baclofen, naltrexone, and sorbitol. Individually, the drugs don't have much effect on the PMP22 gene. But combined, they work to lower how much protein the gene makes.
"How the drug inside the cell reduces expression isn't quite clear yet," says Florian Thomas, director of the Hereditary Neuropathy Center, and founding chair and professor in the department of neurology at Hackensack University Medical Center and Hackensack Meridian School of Medicine in New Jersey (no relation to Robert Thomas, the CMT patient). "By reducing the amount of protein being produced, we hopefully can stabilize the nerves."
In rodents genetically engineered to have the PMP22 gene, the drug reduced protein levels and delayed onset of muscle weakness when given to rats. In another animal study, the drug increased the size of the myelin sheath around nerves in rats.
"Like humans with CMT, one of the problems the animals have is they can't grip things, their grip strength is poor," Solomon says. But when treated with Pharnext's drug, "the grip strength of these animals improves dramatically even over 12 weeks."
Human trials look encouraging, too. But the company ran into a manufacturing issue during a late-stage trial. The drug requires refrigeration, and as a result of temperature changes, crystals formed inside vials containing the high dose of the drug. The study was a double-blind trial, meaning neither the trial participants nor investigators were supposed to know who received the high dose of the drug, who received the low dose, and who received a placebo. In these types of studies, the placebo and experimental drug should look the same so that investigators can't tell them apart. But because only the high dose contained crystals, not the low dose or placebo, regulators said the trial data could be biased.
Pharnext is now conducting a new randomized, double-blind trial to prove that its drug works. The study is recruiting individuals aged 16 through 65 years old with mild to moderate CMT. The company hopes to show that the drug can stop patients' symptoms from worsening, or in the best case scenario, possibly even improve them. The company doesn't think the drug will be able to help people with severe forms of the disease.
"In neurologic disease, you're looking for plasticity, where there's still the possibility of stabilization or reversal," Solomon says. Plasticity refers to the ability of the nervous system to change and adapt in response to stimuli.
Preventing Disability
Allison Moore, a CMT patient and founder and CEO of the Hereditary Neuropathy Foundation, has been following drug development for CMT since she founded the organization in 2001. She says many investigational drugs haven't moved forward because they've shown little success in animals. The fact that Pharnext's drug has made it to a late-stage human trial is promising, she says.
"It's really exciting," Moore says. "There's a chance that if you take the drug early before you're very severe, you'll end up not developing the disease to a level that's super disabling."
CMT has damaged Moore's peroneal nerve, a main nerve in the foot. As a result, she has foot drop, the inability to lift the front part of her foot, and needs to wear leg braces to help her walk. "The idea that you could take this early on and that it could stop progression, that's the hope that we have."
Thomas, the neurologist, says a drug doesn't have to be a cure to have a significant impact on patients. "If I have a CMT patient who's 50 years old, that patient will be more disabled by age 60," he says. "If I can treat that person with a drug, and that person is just as disabled at age 60 as they were at age 50, that's transformative in my mind."
While Robert Thomas says he hasn't noticed a dramatic improvement since he's been on the drug, he does think it's helping. Robert is now in an open-label study, which means he and his health provider are aware that he's receiving the drug.
When the COVID-19 pandemic hit, manufacturing and supply chain disruptions meant that Robert was without the trial drug for two months. When his medication ran out, his legs felt unstable again and walking was harder. "There was a clear distinction between being on and off that medication," he says.
Pharnext's current trial will take about a year and a half to complete. After that, the FDA will decide on whether to approve the drug for CMT patients.
As scientists learn more about the PMP22 gene and the more than 100 other genes that when mutated cause CMT, more precise treatments could be possible. For instance, scientists have used the gene-editing tool CRISPR to correct a CMT-causing mutation in human cells in the lab. The results were published August 16 in the journal Frontiers in Cell and Developmental Biology.
Pharnext is also interested in pursuing genetic treatments for CMT, but in the meantime, repurposed drugs may be the best shot at helping patients until more advanced treatments are available.

"Making Sense of Science" is a monthly podcast that features interviews with leading medical and scientific experts about the latest developments and the big ethical and societal questions they raise. This episode is hosted by science and biotech journalist Emily Mullin, summer editor of the award-winning science outlet Leaps.org.