
How Excessive Regulation Helped Ignite COVID-19's Rampant Spread
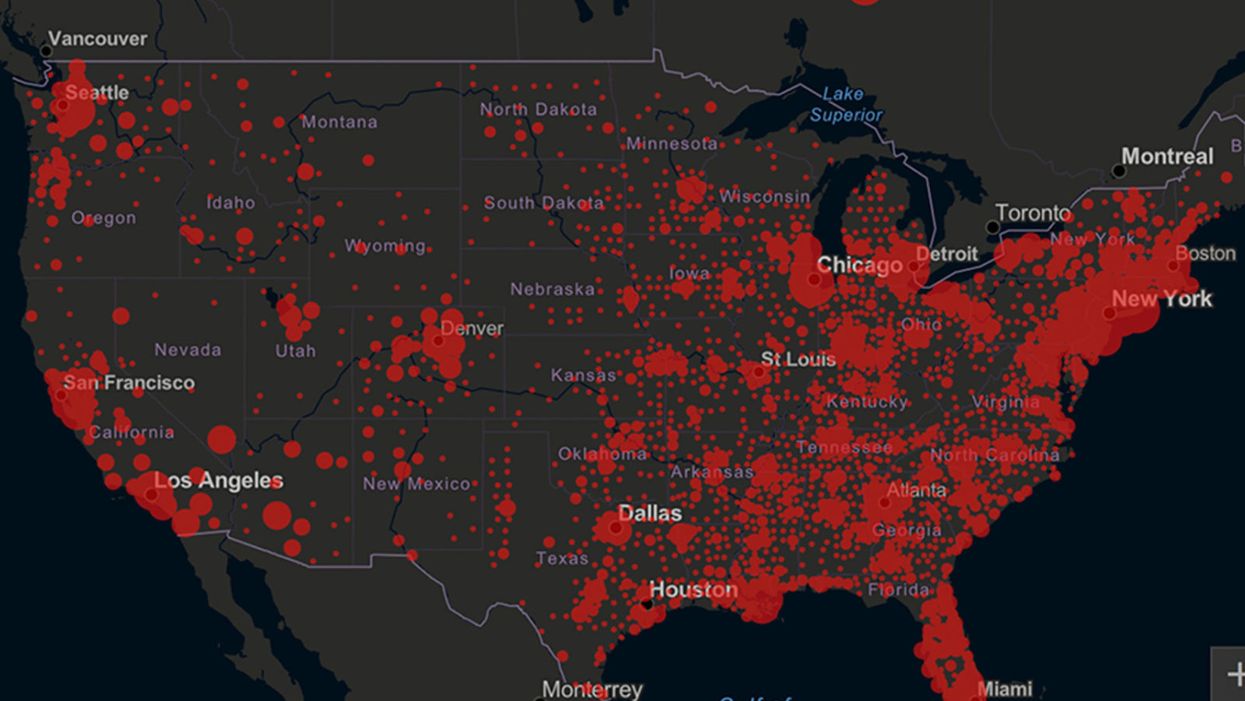
Screenshot of an interactive map of coronavirus cases across the United States, current as of 1:45 p.m. Pacific time on Tuesday, March 31st. Full map accessible at https://coronavirus.jhu.edu/map.html
When historians of the future look back at the 2020 pandemic, the heroic work of Helen Y. Chu, a flu researcher at the University of Washington, will be worthy of recognition.
Chu's team bravely defied the order and conducted the testing anyway.
In late January, Chu was testing nasal swabs for the Seattle Flu Study to monitor influenza spread when she learned of the first case of COVID-19 in Washington state. She deemed it a pressing public health matter to document if and how the illness was spreading locally, so that early containment efforts could succeed. So she sought regulatory approval to adapt the Flu Study to test for the coronavirus, but the federal government denied the request because the original project was funded to study only influenza.
Aware of the urgency, Chu's team bravely defied the order and conducted the testing anyway. Soon they identified a local case in a teenager without any travel history, followed by others. Still, the government tried to shutter their efforts until the outbreak grew dangerous enough to command attention.
Needless testing delays, prompted by excessive regulatory interference, eliminated any chances of curbing the pandemic at its initial stages. Even after Chu went out on a limb to sound alarms, a heavy-handed bureaucracy crushed the nation's ability to roll out early and widespread testing across the country. The Centers for Disease Control and Prevention infamously blundered its own test, while also impeding state and private labs from coming on board, fueling a massive shortage.
The long holdup created "a backlog of testing that needed to be done," says Amesh Adalja, an infectious disease specialist who is a senior scholar at the Johns Hopkins University Center for Health Security.
In a public health crisis, "the ideal situation" would allow the government's test to be "supplanted by private laboratories" without such "a lag in that transition," Adalja says. Only after the eventual release of CDC's test could private industry "begin in earnest" to develop its own versions under the Food and Drug Administration's emergency use authorization.
In a statement, CDC acknowledged that "this process has not gone as smoothly as we would have liked, but there is currently no backlog for testing at CDC."
Now, universities and corporations are in a race against time, playing catch up as the virus continues its relentless spread, also afflicting many health care workers on the front lines.
"Home-testing accessibility is key to preventing further spread of the COVID-19 pandemic."
Hospitals are attempting to add the novel coronavirus to the testing panel of their existent diagnostic machines, which would reduce the results processing time from 48 hours to as little as four hours. Meanwhile, at least four companies announced plans to deliver at-home collection tests to help meet the demand – before a startling injunction by the FDA halted their plans.
Everlywell, an Austin, Texas-based digital health company, had been set to launch online sales of at-home collection kits directly to consumers last week. Scaling up in a matter of days to an initial supply of 30,000 tests, Everlywell collaborated with multiple laboratories where consumers could ship their nasal swab samples overnight, projecting capacity to screen a quarter-million individuals on a weekly basis, says Frank Ong, chief medical and scientific officer.
Secure digital results would have been available online within 48 hours of a sample's arrival at the lab, as well as a telehealth consultation with an independent, board-certified doctor if someone tested positive, for an inclusive $135 cost. The test has a less than 3 percent false-negative rate, Ong says, and in the event of an inadequate self-swab, the lab would not report a conclusive finding. "Home-testing accessibility," he says, "is key to preventing further spread of the COVID-19 pandemic."
But on March 20, the FDA announced restrictions on home collection tests due to concerns about accuracy. The agency did note "the public health value in expanding the availability of COVID-19 testing through safe and accurate tests that may include home collection," while adding that "we are actively working with test developers in this space."
After the restrictions were announced, Everlywell decided to allocate its initial supply of COVID-19 collection kits to hospitals, clinics, nursing homes, and other qualifying health care companies that can commit to no-cost screening of frontline workers and high-risk symptomatic patients. For now, no consumers can order a home-collection test.
"Losing two months is close to disastrous, and that's what we did."
Currently, the U.S. has ramped up to testing an estimated 100,000 people a day, according to Stat News. But 150,000 or more Americans should be tested every day, says Ashish Jha, professor and director of the Harvard Global Health Institute. Due to the dearth of tests, many sick people who suspect they are infected still cannot get confirmation unless they need to be hospitalized.
To give a concrete sense of how far behind we are in testing, consider Palm Beach County, Fla. The state's only drive-thru test center just opened there, requiring an appointment. The center aims to test 750 people per day, but more than 330,000 people have already called to try to book a slot.
"This is such a rapidly moving infection that losing a few days is bad, and losing a couple of weeks is terrible," says Jha, a practicing general internist. "Losing two months is close to disastrous, and that's what we did."
At this point, it will take a long time to fully ramp up. "We are blindfolded," he adds, "and I'd like to take the blindfolds off so we can fight this battle with our eyes wide open."
Better late than never: Yesterday, FDA Commissioner Stephen Hahn said in a statement that the agency has worked with more than 230 test developers and has approved 20 tests since January. An especially notable one was authorized last Friday – 67 days since the country's first known case in Washington state. It's a rapid point-of-care test from medical-device firm Abbott that provides positive results in five minutes and negative results in 13 minutes. Abbott will send 50,000 tests a day to urgent care settings. The first tests are expected to ship tomorrow.
Breakthrough therapies are breaking patients' banks. Key changes could improve access, experts say.
Single-treatment therapies are revolutionizing medicine. But insurers and patients wonder whether they can afford treatment and, if they can, whether the high costs are worthwhile.
CSL Behring’s new gene therapy for hemophilia, Hemgenix, costs $3.5 million for one treatment, but helps the body create substances that allow blood to clot. It appears to be a cure, eliminating the need for other treatments for many years at least.
Likewise, Novartis’s Kymriah mobilizes the body’s immune system to fight B-cell lymphoma, but at a cost $475,000. For patients who respond, it seems to offer years of life without the cancer progressing.
These single-treatment therapies are at the forefront of a new, bold era of medicine. Unfortunately, they also come with new, bold prices that leave insurers and patients wondering whether they can afford treatment and, if they can, whether the high costs are worthwhile.
“Most pharmaceutical leaders are there to improve and save people’s lives,” says Jeremy Levin, chairman and CEO of Ovid Therapeutics, and immediate past chairman of the Biotechnology Innovation Organization. If the therapeutics they develop are too expensive for payers to authorize, patients aren’t helped.
“The right to receive care and the right of pharmaceuticals developers to profit should never be at odds,” Levin stresses. And yet, sometimes they are.
Leigh Turner, executive director of the bioethics program, University of California, Irvine, notes this same tension between drug developers that are “seeking to maximize profits by charging as much as the market will bear for cell and gene therapy products and other medical interventions, and payers trying to control costs while also attempting to provide access to medical products with promising safety and efficacy profiles.”
Why Payers Balk
Health insurers can become skittish around extremely high prices, yet these therapies often accompany significant overall savings. For perspective, the estimated annual treatment cost for hemophilia exceeds $300,000. With Hemgenix, payers would break even after about 12 years.
But, in 12 years, will the patient still have that insurer? Therein lies the rub. U.S. payers, are used to a “pay-as-you-go” model, in which the lifetime costs of therapies typically are shared by multiple payers over many years, as patients change jobs. Single treatment therapeutics eliminate that cost-sharing ability.
"As long as formularies are based on profits to middlemen…Americans’ healthcare costs will continue to skyrocket,” says Patricia Goldsmith, the CEO of CancerCare.
“There is a phenomenally complex, bureaucratic reimbursement system that has grown, layer upon layer, during several decades,” Levin says. As medicine has innovated, payment systems haven’t kept up.
Therefore, biopharma companies begin working with insurance companies and their pharmacy benefit managers (PBMs), which act on an insurer’s behalf to decide which drugs to cover and by how much, early in the drug approval process. Their goal is to make sophisticated new drugs available while still earning a return on their investment.
New Payment Models
Pay-for-performance is one increasingly popular strategy, Turner says. “These models typically link payments to evidence generation and clinically significant outcomes.”
A biotech company called bluebird bio, for example, offers value-based pricing for Zynteglo, a $2.8 million possible cure for the rare blood disorder known as beta thalassaemia. It generally eliminates patients’ need for blood transfusions. The company is so sure it works that it will refund 80 percent of the cost of the therapy if patients need blood transfusions related to that condition within five years of being treated with Zynteglo.
In his February 2023 State of the Union speech, President Biden proposed three pilot programs to reduce drug costs. One of them, the Cell and Gene Therapy Access Model calls on the federal Centers for Medicare & Medicaid Services to establish outcomes-based agreements with manufacturers for certain cell and gene therapies.
A mortgage-style payment system is another, albeit rare, approach. Amortized payments spread the cost of treatments over decades, and let people change employers without losing their healthcare benefits.
Only about 14 percent of all drugs that enter clinical trials are approved by the FDA. Pharma companies, therefore, have an exigent need to earn a profit.
The new payment models that are being discussed aren’t solutions to high prices, says Bill Kramer, senior advisor for health policy at Purchaser Business Group on Health (PBGH), a nonprofit that seeks to lower health care costs. He points out that innovative pricing models, although well-intended, may distract from the real problem of high prices. They are attempts to “soften the blow. The best thing would be to charge a reasonable price to begin with,” he says.
Instead, he proposes making better use of research on cost and clinical effectiveness. The Institute for Clinical and Economic Review (ICER) conducts such research in the U.S., determining whether the benefits of specific drugs justify their proposed prices. ICER is an independent non-profit research institute. Its reports typically assess the degrees of improvement new therapies offer and suggest prices that would reflect that. “Publicizing that data is very important,” Kramer says. “Their results aren’t used to the extent they could and should be.” Pharmaceutical companies tend to price their therapies higher than ICER’s recommendations.
Drug Development Costs Soar
Drug developers have long pointed to the onerous costs of drug development as a reason for high prices.
A 2020 study found the average cost to bring a drug to market exceeded $1.1 billion, while other studies have estimated overall costs as high as $2.6 billion. The development timeframe is about 10 years. That’s because modern therapeutics target precise mechanisms to create better outcomes, but also have high failure rates. Only about 14 percent of all drugs that enter clinical trials are approved by the FDA. Pharma companies, therefore, have an exigent need to earn a profit.
Skewed Incentives Increase Costs
Pricing isn’t solely at the discretion of pharma companies, though. “What patients end up paying has much more to do with their PBMs than the actual price of the drug,” Patricia Goldsmith, CEO, CancerCare, says. Transparency is vital.
PBMs control patients’ access to therapies at three levels, through price negotiations, pricing tiers and pharmacy management.
When negotiating with drug manufacturers, Goldsmith says, “PBMs exchange a preferred spot on a formulary (the insurer’s or healthcare provider’s list of acceptable drugs) for cash-base rebates.” Unfortunately, 25 percent of the time, those rebates are not passed to insurers, according to the PBGH report.
Then, PBMs use pricing tiers to steer patients and physicians to certain drugs. For example, Kramer says, “Sometimes PBMs put a high-cost brand name drug in a preferred tier and a lower-cost competitor in a less preferred, higher-cost tier.” As the PBGH report elaborates, “(PBMs) are incentivized to include the highest-priced drugs…since both manufacturing rebates, as well as the administrative fees they charge…are calculated as a percentage of the drug’s price.
Finally, by steering patients to certain pharmacies, PBMs coordinate patients’ access to treatments, control patients’ out-of-pocket costs and receive management fees from the pharmacies.
Therefore, Goldsmith says, “As long as formularies are based on profits to middlemen…Americans’ healthcare costs will continue to skyrocket.”
Transparency into drug pricing will help curb costs, as will new payment strategies. What will make the most impact, however, may well be the development of a new reimbursement system designed to handle dramatic, breakthrough drugs. As Kramer says, “We need a better system to identify drugs that offer dramatic improvements in clinical care.”

In today's podcast episode, law professor Gaia Bernstein talks about the challenges of keeping control over our thoughts and actions, even when some powerful forces are pushing in the other direction.
Each afternoon, kids walk through my neighborhood, on their way back home from school, and almost all of them are walking alone, staring down at their phones. It's a troubling site. This daily parade of the zombie children just can’t bode well for the future.
That’s one reason I felt like Gaia Bernstein’s new book was talking directly to me. A law professor at Seton Hall, Gaia makes a strong argument that people are so addicted to tech at this point, we need some big, system level changes to social media platforms and other addictive technologies, instead of just blaming the individual and expecting them to fix these issues.
Gaia’s book is called Unwired: Gaining Control Over Addictive Technologies. It’s fascinating and I had a chance to talk with her about it for today’s podcast. At its heart, our conversation is really about how and whether we can maintain control over our thoughts and actions, even when some powerful forces are pushing in the other direction.
Listen on Apple | Listen on Spotify | Listen on Stitcher | Listen on Amazon | Listen on Google
We discuss the idea that, in certain situations, maybe it's not reasonable to expect that we’ll be able to enjoy personal freedom and autonomy. We also talk about how to be a good parent when it sometimes seems like our kids prefer to be raised by their iPads; so-called educational video games that actually don’t have anything to do with education; the root causes of tech addictions for people of all ages; and what kinds of changes we should be supporting.
Gaia is Seton’s Hall’s Technology, Privacy and Policy Professor of Law, as well as Co-Director of the Institute for Privacy Protection, and Co-Director of the Gibbons Institute of Law Science and Technology. She’s the founding director of the Institute for Privacy Protection. She created and spearheaded the Institute’s nationally recognized Outreach Program, which educated parents and students about technology overuse and privacy.
Professor Bernstein's scholarship has been published in leading law reviews including the law reviews of Vanderbilt, Boston College, Boston University, and U.C. Davis. Her work has been selected to the Stanford-Yale Junior Faculty Forum and received extensive media coverage. Gaia joined Seton Hall's faculty in 2004. Before that, she was a fellow at the Engelberg Center of Innovation Law & Policy and at the Information Law Institute of the New York University School of Law. She holds a J.S.D. from the New York University School of Law, an LL.M. from Harvard Law School, and a J.D. from Boston University.
Gaia’s work on this topic is groundbreaking I hope you’ll listen to the conversation and then consider pre-ordering her new book. It comes out on March 28.
